
Trends in Oncology

Why Benchmarking is Now Essential in KRAS Drug Development
The KRAS landscape has changed. As next-generation inhibitors continue to demonstrate clinical impact, the expectations placed on preclinical programmes are increasing. At the centre of this shift is a simple but critical concept. Benchmarking is no longer a differentiator, but a requirement. The Rise of a Clinical Reference Point As therapies such as daraxonrasib advance through clinical development, they establish a new baseline for efficacy. This baseline becomes the reference against which all new KRAS-targeting programmes are evaluated. For preclinical scientists, this fundamentally changes the design of studies. It is no longer sufficient to show that a compound reduces tumor growth in a subset of models. The real question is whether that activity represents meaningful improvement over what is already achievable. Without benchmarking, this question cannot be answered. What Benchmarking Enables Benchmarking provides context and transforms isolated, siloed data into translatable data; the basis on which your decision-making can be trusted. When preclinical results are generated against a known standard, researchers can: Quantify relative efficacy Identify tumor subtypes where a therapy may outperform existing options Understand where resistance patterns differ Evaluate the potential for combination strategies In KRAS-mutant tumors, where heterogeneity is high and resistance is common, this context is essential for making informed decisions. The Importance of Scale and Annotation Not all benchmarking datasets are equal. To be meaningful, benchmarking must be built on three core components: 1. A sufficiently large and diverse model set KRAS mutations span multiple tumor types and molecular contexts, and a robust dataset must reflect this diversity. 2. Deep molecular characterisation Genomics and transcriptomics alone are not enough to fully capture tumor biology. Protein-level and pathway-level data provide additional resolution that is critical for understanding response. 3. Clinical relevance Models must reflect real patient biology, including treatment history and resistance mechanisms. At Champions Oncology, we have generated daraxonrasib response data across more than 50 KRAS-mutant PDX models spanning lung, colorectal, and pancreatic cancers. Each model is fully annotated with clinical and molecular data, enabling direct linkage between response and biology. This is not a conventional screening dataset. It is a translational framework enabling decision-making. Moving Beyond Binary Outcomes Traditional preclinical studies often present results in binary terms. Tumors respond or they do not. While this can be useful at an early stage, it lacks the resolution needed for modern drug development. Benchmarking enables a more nuanced view and instead of asking whether a tumor responds, scientists can ask: How does this response compare to existing therapies? What molecular features distinguish responders from non-responders? Which resistance pathways are likely to emerge? This level of analysis is essential for designing therapies that succeed in the clinic. Implications for Clinical Strategy The value of benchmarking extends beyond the preclinical stage and directly informs clinical development. By linking response data to molecular features, benchmarking datasets can support: Biomarker-driven patient selection Rational combination therapy design Selection of optimal indications Identification of expansion cohorts In a competitive KRAS landscape, these insights provide a critical advantage. A New Standard for Preclinical Research As the field continues to evolve, benchmarking will become a standard component of preclinical workflows. Teams that fail to incorporate benchmarking risk generating data that lacks context and relevance. The future of KRAS research lies in integrated, data-rich approaches that connect biology, pharmacology, and clinical strategy. Benchmarking is the foundation of that future. What Comes Next In the next blog in this series, we examine why many KRAS preclinical models fail to translate into clinical success and how integrated, multi-omic approaches are addressing this gap. Continue exploring KRAS blog posts

KRAS Has Entered a New Era. Why Your Preclinical Strategy Must Change Now
KRAS has long been one of the most important targets in oncology, and for decades it was widely considered “undruggable”. Fortunately, that perception has changed rapidly and today, KRAS-targeted therapies are no longer theoretical. They are clinically validated, increasingly effective, and reshaping how drug development programmes are designed. The transition from early KRAS inhibitors (KRASi) to next-generation compounds represents a fundamental shift in how our field approaches targeted therapy. For preclinical and translational teams, this shift introduces both opportunity and risk. Programs that adapt quickly will be well positioned to succeed, whereas those that rely on outdated models and assumptions may fall behind. A New Standard of Care Is Emerging Recent clinical data for pan-RAS(ON) inhibitors, such as daraxonrasib, signal a turning point in KRAS drug development. As these therapies move toward becoming standard of care, they establish a new benchmark against which all future KRAS-directed agents will be measured. This has immediate implications for preclinical strategies, and it is no longer sufficient to demonstrate activity in isolation. Every new therapy must now be evaluated relative to an evolving clinical reference point. For companies developing therapies in KRAS-mutant indications such as non-small cell lung cancer (NSCLC), colorectal cancer, and pancreatic cancer, the key question has changed. It is no longer “Does this work?” but “Does this outperform or complement what already exists?”. Why Traditional Preclinical Approaches Are No Longer Enough Historically, KRAS drug development has relied heavily on simplified systems such as cell lines or limited in vivo studies. While these approaches played an important role in early discovery, they are insufficient for today’s demands. KRAS biology is highly context dependent and mutation status alone does not determine response. Co-occurring genomic alterations, transcriptional programmes, and protein-level signalling all contribute to therapeutic sensitivity and resistance. As a result, preclinical systems that lack clinical relevance or molecular depth fail to capture the complexity of real tumours. This creates a disconnect between preclinical findings and clinical outcomes, increasing the risk of late-stage failure. The Role of Clinically Relevant Models To address this challenge, there is a growing shift toward clinically relevant models such as patient-derived xenografts (PDXs) with integrated molecular characterisation. These models enable a deeper understanding of tumor biology and provide a more accurate representation of patient response. Importantly, when these models are combined with longitudinal data and treatment history, they allow researchers to study both intrinsic sensitivity and acquired resistance. This is essential in KRAS, where resistance is not an exception but an expected outcome. Benchmarking as the New Foundation In this evolving landscape, benchmarking is becoming the basis of an effective preclinical strategy, and access to datasets that include response to current or emerging standards of care allow scientists to: Identify responder and non-responder populations Understand the molecular drivers of response Design rational combination strategies Prioritise assets with the highest likelihood of success At Champions Oncology, we recognised that benchmarking would become essential in the KRAS space. Our work has focused on building clinically annotated datasets that connect tumor response directly to molecular biology. This enables a level of insight that extends far beyond traditional screening approaches. From Activity to Decision-Making The ultimate goal of preclinical research is not only to generate data, but to inform your decision making. In KRAS drug development, the stakes are high. Clinical trials are expensive, timelines are long, and the competitive landscape is rapidly evolving. Preclinical data must therefore do more than demonstrate activity. It must guide: Patient selection strategies Combination therapy design Clinical trial structure Pipeline prioritisation This requires a shift from descriptive models to more predictive, translational systems. Looking Ahead KRAS has entered a new era. The science is advancing, the clinical landscape is evolving, and expectations are rising across the industry. For preclinical teams, this is a moment to reassess how studies are designed, how models are selected, and how data are interpreted. The organisations that lead in this space will be those that align their strategy with the realities of the current landscape, not the assumptions of the past. In the next blog in this series, we explore why benchmarking is no longer optional in KRAS research and how it is reshaping preclinical decision-making. Poster: Molecular determinants of sensitivity and resistance to the pan-RAS(ON) inhibitor daraxonrasib
Daraxonrasib Is Rewriting the Standard of Care. Here's Why Preclinical Benchmarking Matters Now
A Standing Ovation and a New Standard of Care At the 2026 ASCO Annual Meeting, the RASolute 302 Phase 3 trial delivered what many are calling the most significant advance in pancreatic cancer treatment in decades. Daraxonrasib, a first-in-class oral RAS(ON) multi-selective inhibitor developed by Revolution Medicines, nearly doubled overall survival (OS) in previously treated metastatic pancreatic ductal adenocarcinoma (PDAC), achieving a median OS of 13.2 months compared to 6.7 months with standard chemotherapy (HR 0.40; p<0.0001). Progression-free survival followed the same trajectory: 7.3 months versus 3.5 months. The results, published simultaneously in The New England Journal of Medicine, drew a standing ovation during the plenary session. ASCO's chief medical officer called it "a grand slam," and the ASCO-selected commentator described starting to cry in clinic after seeing the data. This is no longer a drug to watch. Daraxonrasib is poised to become the new standard of care for second-line metastatic PDAC, with first-line combination trials already underway. For drug developers working in KRAS-driven cancers, the question is no longer whether daraxonrasib will reshape the treatment landscape, but how fast, and whether your preclinical program is ready. Why Benchmarking Against Daraxonrasib Is Now Essential More than 90% of pancreatic cancers harbor KRAS mutations, making it one of the most RAS-addicted tumor types in oncology. As daraxonrasib transitions from investigational therapy to standard of care, it will become the benchmark against which new agents, novel combinations, and next-generation therapies are measured. This shift has immediate implications for preclinical strategy. Scientists developing therapies in KRAS-mutant NSCLC, colorectal cancer, and PDAC need access to clinically annotated models with existing daraxonrasib response data to design meaningful benchmarking and combination studies. Without this data, preclinical programs risk testing against an outdated treatment landscape. Yet despite the urgency, a significant gap exists. Most contract research organizations and preclinical providers do not have daraxonrasib response data, the KRAS-mutant patient-derived xenograft (PDX) models needed to generate it, or the multi-omic depth required to interpret results in a translational context. Champions Oncology Has Already Built the Dataset At Champions Oncology, we recognized early that daraxonrasib would become a defining compound in the KRAS space. That is why we have already screened over 50 KRAS-mutant PDX models across NSCLC, colorectal cancer, and PDAC, generating a unique and comprehensive daraxonrasib benchmarking dataset that is available today. Figure 1: Tumor response and genomic landscape of KRAS-mutant PDX models treated with Daraxonrasib (RMC-6236) This is not a standard drug screen. Every model in the cohort is fully annotated with clinical history, treatment status, and deep molecular profiling, including whole exome sequencing, RNA sequencing, and integrated genomic analysis. The resulting waterfall plots and genomic annotation panels allow scientists to rapidly identify responder and non-responder models matched to their therapeutic hypothesis, and to understand the molecular context driving each outcome. Understanding what drives resistance. Differential gene expression analysis of responding versus non-responding KRAS G12D-mutant PDAC models has identified transcriptional programs associated with primary resistance to daraxonrasib, providing actionable insights for biomarker development and patient selection strategies. Modeling acquired resistance in real time. Because secondary resistance is an inevitable feature of targeted therapy, we have developed acquired resistance models under continuous daraxonrasib treatment pressure. Models CTG-2473 (pancreatic, KRAS G12D) and CTG-0068 (colorectal, KRAS G12D) both demonstrated initial tumor regression followed by regrowth, recapitulating the resistance patterns expected in patients. These models create a powerful translational framework for testing next-line agents, rational combination strategies, and treatment sequencing approaches. Going deeper with multi-omic prediction. Champions' Pharmaco-Pheno-Multiomic (PPMO) integration platform takes this further by layering whole-cell proteomics, cell surface proteomics, genomics, and transcriptomics across 56 KRAS-mutant PDX models treated with daraxonrasib. The resulting computational model achieves 80% prediction accuracy for drug response, substantially outperforming RNA-only approaches, which plateau at approximately 60%. This level of biological resolution is critical for identifying the molecular determinants of sensitivity and resistance that standard biomarker panels miss. Download the Full Poster These findings were first presented at AACR 2026 (Poster 1884): Molecular Determinants of Sensitivity and Resistance to the Pan-RAS(ON) Inhibitor Daraxonrasib (RMC-6236) Across KRAS-Mutant Patient-Derived Models. For the complete dataset, including response profiles, genomic annotations, and resistance modeling data, download the full poster here. For additional context, read our companion blog: Understanding Sensitivity and Resistance to Pan-RAS(ON) Inhibition Across KRAS-Mutant Tumors. The Landscape Is Shifting. Let's Talk About Your Program. Daraxonrasib is changing how KRAS-targeted therapies will be developed and evaluated. Whether you are benchmarking a novel compound against an emerging standard of care, designing combination strategies, or investigating mechanisms of primary and acquired resistance, we have the models, the data, and the translational depth to support your program. Contact us today to discuss your study
Scaling Chem-Seq and Functional-Seq to Decode Drug and Genetic Perturbations in Patient-Derived Organoids
Large-scale drug discovery increasingly depends on understanding mechanism, not just measuring viability. While high-throughput screening has traditionally relied on simple 2D systems, these models often fail to capture the biological complexity of human tumors or provide the mechanistic depth needed to make confident development decisions. At the same time, transcriptomic approaches have historically been too low-throughput to support large compound libraries. In our AACR 2026 poster, we introduced scalable Chem-Seq and Functional-Seq platforms designed to close this gap. By combining high-throughput transcriptomic profiling with patient-derived organoid models, we can systematically map chemical and genetic perturbations at scale while preserving clinically relevant tumor biology. Why transcriptomic profiling matters in drug discovery Phenotypic screening alone rarely explains why a compound works or fails. Two agents may produce similar viability effects for completely different reasons, while others may appear inactive despite engaging their intended targets. Transcriptomic profiling addresses this challenge by capturing pathway-level responses and signatures of mechanism of action. However, traditional RNA-seq workflows are difficult to scale. Sample preparation, library generation, and sequencing cost all become limiting factors. Our goal was to develop a workflow that preserves transcriptomic resolution while enabling tens of thousands of perturbations per week in relevant cancer models. A scalable Chem-Seq workflow built for throughput Our Chem-Seq platform is built around DRUG-seq™, a workflow optimized for bulk mRNA profiling directly from cancer cell lines or PDX-derived organoids in 384-well plates. Cells or organoids are plated, cultured, and treated with small-molecule compounds. Following treatment, a single-step lysis and barcoded oligo-dT capture allows mRNA from each well to be uniquely indexed in situ. After reverse transcription, all wells are pooled, amplified, and processed through standard Illumina library preparation workflows. Libraries are sequenced to a depth sufficient to capture robust transcriptional signatures while maintaining scalability. This approach supports roughly 50,000 perturbations per week, spanning small molecules, and siRNAs. The key advantage is efficiency. By eliminating many traditional bottlenecks, Chem-Seq enables rapid, reproducible transcriptomic profiling at a scale that aligns with modern screening demands. From raw reads to mechanism-resolved insights Chem-Seq data are processed to generate gene-by-barcode matrices, followed by normalization, batch correction, and dimensionality reduction. Principal component analysis and UMAP visualizations enable rapid triage of perturbation effects, while differential expression and pathway analyses reveal mechanism-associated signatures. To further contextualize results, Chem-Seq signatures are compared against large public perturbation datasets using connectivity mapping approaches. This allows unknown compounds to be matched to known drugs, siRNA knockdowns, or CRISPR perturbations based on shared transcriptional responses, providing early insight into mechanism of action and potential off-target effects. Anchoring chemistry with Functional-Seq While chemical perturbations provide rich information, interpreting their biological meaning is often strengthened by genetic context. Functional-Seq integrates siRNA-mediated knockdown of drug targets directly into the same transcriptomic framework. In this study, Functional-Seq was applied in non-small cell lung cancer PDX organoid models to generate loss-of-function signatures for selected targets. These genetic signatures serve as anchors for interpreting Chem-Seq profiles, enabling direct comparison between pharmacologic inhibition and genetic suppression of the same pathway. Where chemical and genetic perturbations converge transcriptionally, confidence in on-target activity is strengthened. Where they diverge, additional biology, compensatory signaling, or off-target effects can be explored. What the platform reveals in patient-derived organoids Using patient-derived organoids preserves key aspects of tumor heterogeneity and 3D architecture that are often lost in standard cell lines. Across the presented NSCLC PDX organoid models, Chem-Seq generated highly reproducible transcriptional signatures. Compounds targeting shared pathways clustered together, even when structurally distinct, while strongly cytotoxic agents formed separate transcriptional groups. Dose-response profiling added an additional layer of insight. Increasing compound concentration revealed clear transitions between pathway-selective signatures and broader cytotoxic stress responses. This distinction is critical for prioritizing compounds that modulate their intended targets without inducing nonspecific toxicity. Functional-Seq knockdowns recapitulated chemical inhibition for several pathways, demonstrating strong cross-method concordance at the gene and pathway levels. In contrast, targets that failed to produce robust genetic or pharmacologic signatures highlighted limitations related to knockdown efficiency or pathway redundancy. Why integrated Chem-Seq and Functional-Seq matter Together, Chem-Seq and Functional-Seq provide a scalable, mechanism-resolved framework for drug discovery in clinically relevant models. This integrated approach supports: Mechanism confirmation and target engagement assessment Rapid prioritization of compounds with clean, pathway-specific signatures Early identification of off-target or cytotoxic liabilities Improved interpretation of phenotypic screening data Because the platform is compatible with large compound libraries and patient-derived organoids, it bridges the gap between discovery-scale screening and translational relevance. Looking ahead Following validation, this workflow is designed to scale further. Large-scale screening across diverse PDX organoid models and standard-of-care comparators will expand the reference space of transcriptomic signatures. Combined with machine-learning and generative AI approaches, these datasets can be used to predict compound response, guide structure-activity relationships, and even design novel compounds optimized for desired transcriptional outcomes. By integrating high-throughput transcriptomics, genetic perturbation anchoring, and patient-derived 3D biology, Chem-Seq and Functional-Seq offer a powerful foundation for next-generation oncology drug discovery and precision medicine. Want to explore the full data? This blog highlights the overall strategy and key insights, but the complete poster includes detailed workflows, clustering analyses, concordance metrics, and pathway-level results. Download the AACR 2026 poster to explore the full Chem-Seq and Functional-Seq dataset and figures.
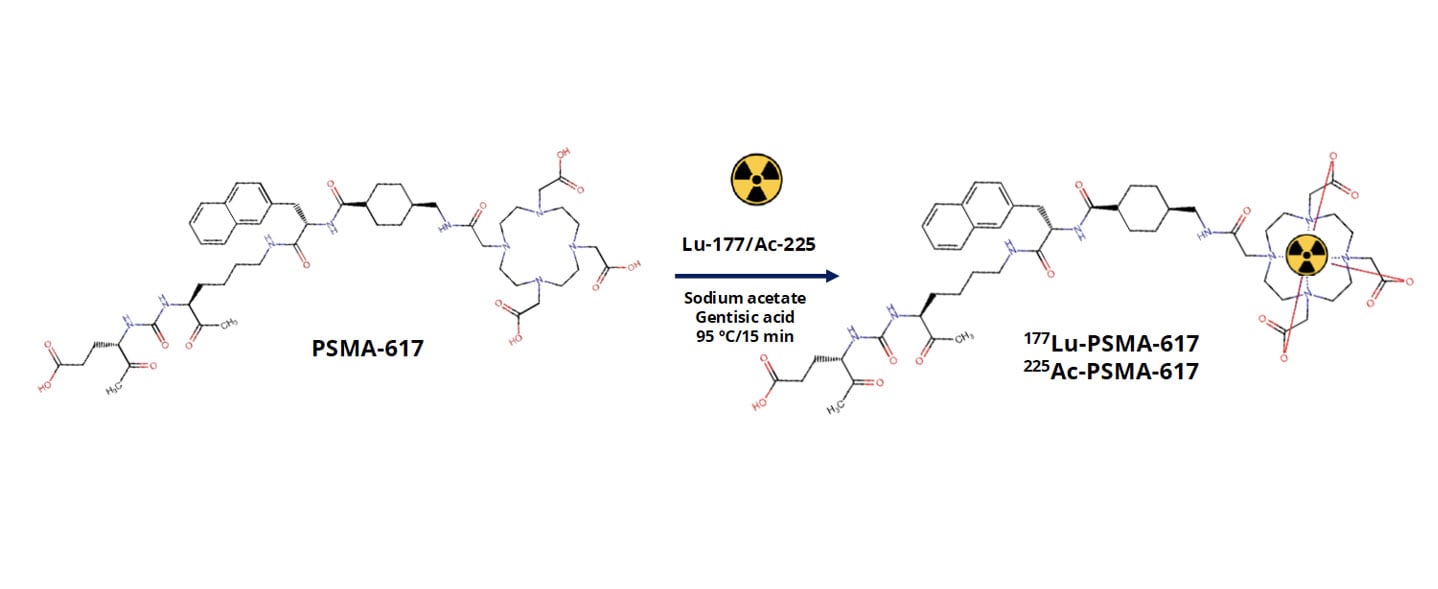
How Preclinical PDX Studies Help Guide PSMA Radioligand Development
Metastatic castration-resistant prostate cancer, or mCRPC, remains a major clinical challenge. While prostate-specific membrane antigen, PSMA, has emerged as a highly promising therapeutic target, differences in tumor biology, target expression, and radiopharmaceutical behavior continue to influence clinical outcomes. For PSMA-targeted radioligand therapies to reach their full potential, developers need preclinical models that reliably capture both tumor complexity and biodistribution behavior in vivo. At AACR 2026, we presented data showing how patientderived xenograft (PDX) models can be used to evaluate PSMA-targeted radiopharmaceuticals at multiple levels. This included radiochemistry quality, biodistribution, and tumor selectivity. The work focused on two clinically relevant agents, [177Lu]LuPSMA617 and [225Ac]AcPSMA617, tested across a panel of mCRPC PDX models with variable PSMA expression. Why PDX models are critical for radiopharmaceutical development Radiopharmaceutical therapies rely on far more than target binding alone. Successful translation depends on tumor uptake, retention, clearance from healthy tissues, and therapeutic index. Traditional cell line models fail to capture the architectural and microenvironmental features that shape these processes in patients. PDX models preserve the genetic heterogeneity and tissue organization of the original human tumor. This makes them particularly well suited for studying radioligand distribution and efficacy in vivo. In the context of PSMA-targeted therapies, PDX models allow us to directly evaluate how differences in PSMA expression and tumor biology influence radiotracer uptake in clinically relevant tumor models. Designing a clinically relevant PSMA radiopharmaceutical study In this study, we evaluated PSMA617 labeled with either lutetium177 or actinium225 across multiple prostate cancer PDX models representing metastatic, castration-resistant disease. The models spanned a range of PSMA expression levels, enabling us to assess how target expression directly relates to biodistribution. We first confirmed robust radiochemical performance. PSMA617 was efficiently labeled with both isotopes, achieving high radiochemical purity that met stringent quality standards. Establishing radiochemistry consistency is a critical first step before moving into in vivo evaluation, particularly for comparative studies involving different isotopes. How biodistribution studies inform target specificity and safety Following radiotracer administration, we conducted biodistribution analyses at defined time points to quantify uptake across tumors and major organs. Rather than relying on a single model, we assessed multiple PDXs to capture biologically driven variability. Across the panel, tumor uptake correlated strongly with PSMA expression levels. Models with high PSMA expression showed selective and robust radiotracer accumulation, while low-PSMA models demonstrated minimal uptake. This clear relationship confirms biological predictability and reinforces the value of molecularly annotated PDX panels when evaluating PSMA-targeted therapies. Importantly, although both [177Lu]LuPSMA617 and [225Ac]AcPSMA617 demonstrated tumor targeting, differences emerged in tumor-to-normal tissue ratios. These distinctions highlight why side-by-side preclinical evaluation is essential when selecting isotopes and dosing strategies for clinical development. Comparing beta- and alpha-emitting PSMA therapies One of the most important questions in PSMA radiopharmaceutical development is how different isotopes behave in vivo. Lutetium177 and actinium225 differ in emission properties, tissue penetration, and potential toxicity profiles. Our comparative biodistribution analyses showed that while both agents localize to PSMA-expressing tumors, [177Lu]LuPSMA617 demonstrated more favorable tumortotissue ratios across several organs. These findings provide early insight into how isotope choice may influence therapeutic window and tolerability. While alpha-emitters remain highly compelling for their potent cytotoxicity, data like this emphasize the importance of rigorous preclinical evaluation in clinically relevant models before advancing treatment strategies. Why this approach matters for radiopharmaceutical pipelines Radiopharmaceutical development is inherently multidisciplinary, integrating chemistry, biology, imaging, and therapeutic evaluation. PDX models provide a unifying platform where all of these components can be assessed in context. Using well-characterized mCRPC PDXs allows us to: Link PSMA expression directly to in vivo uptake Compare isotopes in clinically relevant tumor models Evaluate tumor selectivity alongside therapeutic response Reduce translational uncertainty before clinical studies This kind of integrated preclinical strategy is especially valuable as PSMA-targeted therapies continue to expand into earlier disease settings and combination regimens. Want to see the full dataset? This blog highlights key findings, but the full study includes detailed radiochemistry validation, biodistribution data, tumor-to-tissue ratios, and efficacy results across multiple PDX models. Download our AACR 2026 poster to explore the complete results and analyses.
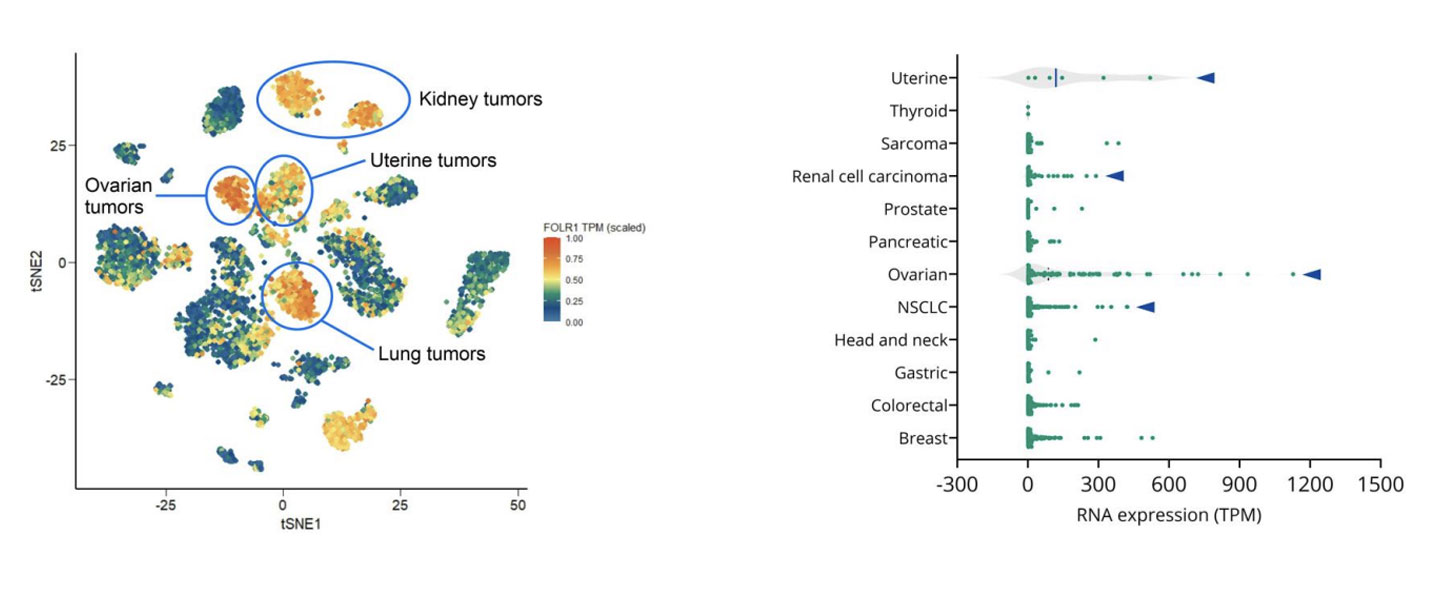
How to Use PDX Models to Advance Folate Receptor-Alpha ADC Development in Ovarian Cancer
Epithelial ovarian cancer remains one of the deadliest gynecologic malignancies. Although many patients initially respond well to platinum-based chemotherapy, relapse is common, and outcomes for platinum-resistant disease have seen little improvement over the past decade. With limited treatment options and a shortage of robust predictive biomarkers, there is a clear need for more precise therapeutic strategies that can better match patients to targeted therapies. One promising area of progress is antibody-drug conjugates, or ADCs. These therapies are designed to deliver potent cytotoxic agents directly to tumor cells by targeting tumorassociated antigens. In ovarian cancer, folate receptoralpha, or FRα, has emerged as an especially compelling target due to its high expression in ovarian tumors and comparatively limited expression in normal tissues. At AACR 2026, we shared new data showing how patient-derived xenograft, or PDX, models can be used to evaluate FRα-targeted ADCs, explore variability in response, and begin to uncover potential mechanisms of resistance. This work highlights how clinically relevant PDX models, combined with molecular profiling, can support smarter ADC development decisions earlier in the pipeline. Why FRα is an attractive ADC target in ovarian cancer FRα is a transmembrane glycoprotein involved in folate transport and cellular metabolism. While healthy tissues show limited FRα expression, ovarian tumors, particularly high-grade serous carcinomas, often express it at high levels. This contrast makes FRα well suited for ADC approaches that aim to maximize tumor-specific activity while minimizing offtarget toxicity. Mirvetuximab soravtansine-gynx, marketed as Elahere®, is an FRα-targeted ADC recently approved for patients with FRα-positive, platinum-resistant ovarian cancer. However, clinical experience has shown that FRα expression alone does not reliably predict which patients will respond to treatment. Understanding this response heterogeneity is one of the key challenges in optimizing ADC development. How we select the right PDX models to reflect patient biology To address this challenge, we leveraged our gynecologic cancer PDX collection, which includes models derived from both treatment-naïve and heavily pretreated ovarian cancer patients. Using integrated RNA sequencing and immunohistochemistry, we assessed FRα expression across ovarian PDXs and compared these patterns with patient data. Consistent with clinical observations, ovarian cancer PDXs showed some of the highest FOLR1 expression levels across tumor types. Importantly, RNAbased expression data strongly correlated with membrane-specific FRα protein expression. This confirms that multiple complementary methods can be used to select biologically appropriate models for FRαtargeted ADC screening. Rather than relying on a single cutoff, this approach allows us to build PDX cohorts that better reflect the biological diversity seen in the clinic. What PDX efficacy studies reveal about ADC response Selected ovarian PDX models were treated with mirvetuximab soravtansine using a clinically relevant dosing schedule. As expected, models lacking FRα expression did not respond to treatment, reinforcing the importance of FRα as a prerequisite for activity. Among FRα-positive models, however, responses varied widely. Some tumors showed meaningful growth inhibition, while others demonstrated limited or no response despite high FRα expression. This mirrors what is seen in the clinic, where only a subset of FRαpositive patients achieves durable benefit from treatment. These results highlight a critical lesson for ADC development. Target expression is necessary, but it is rarely sufficient on its own. Using molecular analysis to explore resistance mechanisms To begin understanding why FRα-positive tumors respond differently, we compared responding and non-responding PDX models using transcriptomic and pathway-level analyses. Rather than focusing on single genes, we looked at broader biological processes that may influence sensitivity or resistance to ADC therapy. Clear differences emerged between sensitive and resistant tumors. In particular, pathways related to intracellular transport and drug efflux were enriched in non-responding models. Several of these pathways involve ATP-binding cassette transporters, which are well known for their role in multidrug resistance. While exploratory, these findings suggest potential mechanisms by which tumor cells may evade ADC-delivered payloads, even when the target antigen is present. This type of insight can help guide biomarker development and inform rational combination strategies. How we model acquired resistance using PDXs Primary resistance is only part of the challenge. In many patients, response to ADCs is followed by progression under treatment. To study this, we are actively generating PDX models from patients who initially responded to ADC therapy and later relapsed. In the AACR study, one such PDX was derived from a patient who had responded to mirvetuximab soravtansine before progressing. When re-challenged in vivo, this model showed resistance to treatment, suggesting that clinically acquired resistance mechanisms can be preserved in PDXs. Models like this provide powerful tools for studying resistance biology and evaluating nextgeneration therapies designed to overcome it. Why PDXs matter for ADC development Taken together, these findings show how PDX models can support ADC development beyond basic efficacy screening. When combined with molecular and bioinformatics analyses, they can help: Improve patient stratification beyond single biomarkers Reveal biological drivers of heterogeneous response Enable investigation of intrinsic and acquired resistance Support the development of next-generation ADCs and combination approaches As ADC pipelines continue to expand, integrating clinically representative PDX models early in development can improve translation and, ultimately, patient outcomes. Want to explore the full data? This blog highlights key insights, but the full dataset includes detailed efficacy results, pathway analyses, and figures that provide additional depth. Download our AACR 2026 poster to explore the complete results and analyses.
Understanding Sensitivity and Resistance to Pan-RAS(ON) Inhibition Across KRAS-Mutant Tumors
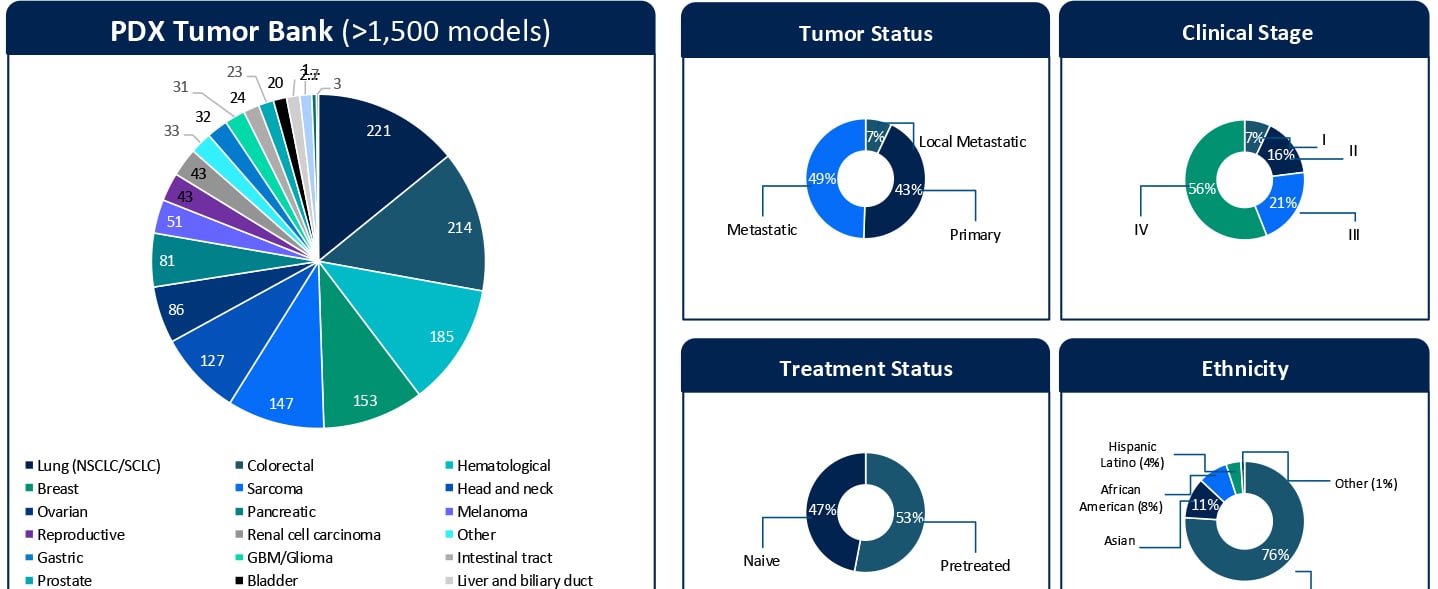
Understanding Sensitivity and Resistance to Pan-RAS(ON) Inhibition Across KRAS-Mutant Tumors KRAStargeted therapies are entering a new phase of clinical relevance. Daraxonrasib, a next-generation pan-RAS(ON) inhibitor designed to target a broad spectrum of KRAS G12X mutations, has shown impressive clinical activity. In a recent Phase III RASolute 302 trial for second-line metastatic pancreatic ductal adenocarcinoma (PDAC), daraxonrasib (RMC-6236) nearly doubled the median overall survival for patients to approximately 13.2 months, compared with 6.7 months for standard chemotherapy.1 These results highlight the rapid progress in this space. At the same time, clinical development is expanding KRAS-targeted therapies into earlier lines of therapy. Daraxonrasib is currently being evaluated in first-line settings, both as monotherapy and in combination approaches. While both clinical and preclinical data demonstrate activity across multiple tumor types, variability in response and the emergence of resistance remain key challenges that limit durable benefit. Additionally, the molecular determinants that define sensitivity, primary resistance, and acquired resistance remain incompletely understood. Addressing these gaps in our understanding of the molecular drivers of sensitivity and resistance is critical not only for patient selection but also for informing rational combination and sequencing strategies in an evolving treatment landscape. A Translational Platform to Study KRAS Biology in Context To better understand these response patterns, this study leveraged Champions Oncology’s low-passage, clinically annotated Patient-Derived Xenograft (PDX) Tumor Bank. With more than 1,500 models and extensive molecular and treatment history annotation, this platform captures both tumor heterogeneity and clinically relevant treatment context. Importantly, approximately half of the models represent pretreated disease, reflecting real-world patient populations. The translational relevance of this platform is supported by prior clinical correlation analyses performed using Champions’ low-passage PDX models. In a study by Izumchenko et al., treatment responses observed in these models were directly compared with clinical outcomes in matched patients across 92 individuals and 129 therapies.2 Strong concordance between the PDX and patient response was observed, with a positive predictive value of approximately 85% and a negative predictive value of approximately 91%. These findings establish Champions’ low-passage PDX models both as a tool for biological exploration and a clinically validated system for informing translational and drug development decisions. In this study, more than 50 KRAS-mutant PDX models were screened across non-small cell lung cancer, colorectal cancer, and pancreatic cancer (PDAC). The cohort included diverse KRAS alleles, co-occurring genomic alterations, and varying treatment histories, which allowed treatment response to be evaluated within a realistic molecular and clinical framework. Heterogeneous Responses Reflect Tumor-Specific Context Daraxonrasib treatment resulted in a broad spectrum of responses, ranging from strong disease regressions to progressive disease. Response variability was not solely explained by KRAS mutation status alone but was influenced by the broader genomic context of each tumor. For example, alterations such as MYC amplification were observed in both responding and non-responding models, suggesting that primary resistance is context-dependent and likely driven by multiple interacting genomic factors, rather than single genomic features. These findings highlight the importance of distinguishing between intrinsic resistance mechanisms and those that emerge under therapeutic pressure. Probing Primary Resistance in KRAS G12D Pancreatic Models Given the clinical relevance of KRAS G12D in pancreatic cancer, a focused analysis was performed in this subset. Differential gene expression analysis comparing responders and non-responders identified candidate markers associated with response, including MSLN and CYP24A1. While these findings are exploratory and based on a limited sample set, they provide an initial framework for stratifying tumors and generating hypotheses around mechanisms of intrinsic resistance. Ongoing work is expanding these analyses through deeper multi-omic integration, including whole exome sequencing (WES), RNA sequencing, proteomics, phosphoproteomics, and surface proteomics. These datasets will be interpreted in the context of Champions’ clinically annotated models, which include detailed treatment histories, to better understand the biological drivers of response and resistance and to support more robust biomarker development. Modeling Acquired Resistance Under Continuous Drug Pressure Selected PDX models were treated continuously beyond their initial response to investigate how resistance evolves over time. A particularly illustrative example is model CTG-2473, a KRAS G12D pancreatic cancer model derived from a tumor collected after first-line gemcitabine treatment. This model reflects a clinically relevant treatment sequence where patients, following standard chemotherapy, would be transitioned to KRAS-targeted therapies such as daraxonrasib. Therefore, treatment of CTG-2473 with daraxonrasib can model response and resistance in a post-chemotherapy setting that mirrors emerging clinical practice. Under continuous treatment pressure, this model demonstrated initial tumor regression followed by relapse, which is consistent with the development of acquired resistance observed in patients. This creates a powerful framework for translational studies. Experiments can be designed in the parental setting to evaluate therapeutic switching upon relapseto assess next-line agents or rational combination strategies. In parallel, Champions Oncology is actively developing a fully resistant subline through serial passaging under sustained drug pressure. This will enable deeper molecular characterization of acquired resistance mechanisms and provide a robust system for testing strategies for overcoming resistance. Together, these models establish a framework not only to observe resistance but also to actively model clinically relevant treatment sequencing scenarios. Why Clinically Annotated PDX Models Matter A defining strength of this work is the integration of molecular profiling, treatment response, and detailed clinical annotation within a single experimental system. Unlike conventional preclinical models, Champions’ PDX platform enables analysis of tumor behavior in the context of prior therapies, disease progression, and patient-specific treatment history. This is particularly important for targeted therapies such as KRAS inhibitors, where response and resistance are shaped by both molecular context and prior treatment. When combined with multi-omic datasets, including WES, RNA sequencing, proteomics, phosphoproteomics, and surface proteomics, these clinically annotated models provide a systems-level understanding of tumor biology. This supports the identification of candidate biomarkers and the development of mechanistically informed combination and sequencing strategies. Importantly, this integrated approach allows researchers to move beyond static response measurements tto capture how tumors dynamically adapt under therapeutic pressure and how these adaptations can be therapeutically targeted. From Mechanism to Strategy Taken together, these data demonstrate that response to pan-RAS(ON) inhibition is shaped by tumor-specific molecular context and dynamic adaptation under treatment. Understanding both primary and acquired resistance will be essential for translating KRAS-targeted therapies into durable clinical benefit. The full dataset, including detailed response profiles, genomic annotations, and resistance modeling, is presented in the accompanying poster. Download the Full Poster To explore the complete results, figures, and molecular analyses, including responder and non-responder comparisons and acquired resistance models, download the full poster here. References Daraxonrasib Demonstrates Unprecedented Overall Survival Benefit in Pivotal Phase 3 RASolute 302 Clinical Trial in Patients with Metastatic Pancreatic Cancer | Revolution Medicines [Internet]. Revolution Medicines. 2026. Available from: https://ir.revmed.com/news-releases/news-release-details/daraxonrasib-demonstrates-unprecedented-overall-survival-benefit Izumchenko E, Paz K, Ciznadija D, Sloma I, Katz A, Vasquez-Dunddel D, et al. Patient-derived xenografts effectively capture responses to oncology therapy in a heterogeneous cohort of patients with solid tumors. Annals of Oncology. 2017 Oct;28(10):2595–605.
Clinically Relevant 3D Tumor Models for HER2-Targeted ADC Development
Expanding the Preclinical Toolbox with a Novel Patient-Derived Organoid Platform The human epidermal growth factor receptor 2 (HER2) plays a central role in regulating cell growth, division, and survival. When overexpressed, it acts as an "on switch", driving accelerated cell growth and uncontrolled tumor spread. This biology underlies the aggressive behavior observed in HER2-positive cancers, which is associated with higher recurrence rates and poorer patient outcomes. While genomic changes in HER2 are most frequently discussed in the context of breast cancer, HER2 alterations are also found in non-small cell lung, ovarian, colorectal, and pancreatic cancers. HER2-Targeted ADCs in Cancer Therapy HER2-targeted therapies have significantly changed the treatment landscape and brought new hope and better outcomes for patients. Among them, HER2-targeted antibody-drug conjugates (ADCs) have emerged as a particularly promising approach, combining the specificity of antibodies with the potency of cytotoxic payloads. These agents enable targeted delivery directly to HER2-expressing tumor cells while limiting off-target toxicity. FDA approved drugs in this class include trastuzumab emtansine (T-DM1, Kadcyla®) and trastuzumab deruxtecan (T-DXd, Enhertu®), both of which have demonstrated meaningful clinical responses (Figure 1). A growing number of next-generation HER2 ADCs are now in development. Figure 1. FDA approved HER2-targeted ADCs trastuzumab emtansine (T-DM1, Kadcyla®) and trastuzumab deruxtecan (T-DXd, Enhertu®). Figure adapted from Joubert et al1. As ADCs become an increasingly important therapeutic modality, the limitations of conventional preclinical models have become magnified. Drug developers need better, more physiologically relevant and predictive preclinical models to support ADC development, ones capable of reproducing tumor architecture, heterogeneity, and treatment response. Limitations of Traditional 2D Preclinical Models for ADC Evaluation Despite their widespread use, traditional 2D cell culture models have well-recognized drawbacks with respect to evaluating targeted therapies. They often fail to capture key biological features of tumors and inadequately replicate the complex tumor microenvironment (TME) that influences drug response in patients. Target accessibility, receptor density, internalization kinetics, and payload penetration are all influenced by three-dimensional tumor structure, features which are largely absent in 2D cultures. As a result, 2D assays may overestimate drug potency or fail to distinguish on-target activity from nonspecific cytotoxicity, reducing their predictive value for clinical translation. In contrast, growing tumor cells as 3D organoids allows for a spatially organized structure that more closely resembles the in vivo microenvironment and architecture of patient tumors. Champions has developed a scalable 3D screening platform based on HER2-positive patient-derived xenograft organoids (PDXOs). Derived from well-characterized PDX models, our ex vivo 3D organoids maintain clinically translatable HER2 expression, providing a relevant and predictive platform for evaluating the efficacy of HER2-targeted ADCs. Validation of HER2-Positive PDXO Models HER2-positive ex vivo PDXO breast cancer models generated from our TumorGraft3D (CTG-3D) platform were evaluated for HER2 expression as well as functional responses to the clinically approved HER2-targeted ADCs, trastuzumab emtansine (T-DM1) and trastuzumab deruxtecan (T-Dxd). The workflow is shown in Figure 2. Figure 2. Workflow overview to evaluate HER2-targeted ADCs using breast cancer PDXO models. HER2 Expression Immunohistochemical (IHC) analysis of PDXO tissue blocks demonstrated complete and intense membrane staining (HER2 score 3+) across HER2-positive models. Consistent with these findings, flow cytometric analysis of enzyme-dissociated organoids identified HER2-positivity in 61.8% to 90.1% of cells. In contrast, the HER2-negative control PDXO model showed no detectable HER2 staining by IHC and negligible HER2 expression by flow cytometry, confirming assay specificity and the absence of nonspecific background signal. A subset of the results is shown in Figure 3. Figure 3. HER2 expression assessed by IHC and flow cytometry analysis. A) HER2-positive PDXO and B) HER2-negative PDXO model. Together, the IHC staining and flow cytometry results are consistent with the clinical annotation and indicate that HER2 expression is maintained relatively homogeneously in breast cancer PDXO models. Response to HER2-Targeted ADCs After confirmation of HER2 expression, the PDXO models were evaluated for their response to two clinically approved HER2-targeted ADCs, trastuzumab deruxtecan (T-DXd) and trastuzumab emtansine (T-DM1). Both ADCs were tested against HER2-positive and HER2-negative breast cancer PDXOs and reference breast cancer cell lines. In addition to intact ADCs, corresponding cytotoxic payloads (Exatecan and DM1), naked antibody (trastuzumab), and—for T-DXd—an isotype-payload conjugate (IgG-DXd) were evaluated to distinguish potency, selectivity, and target-dependent activity. Drug response was measured using the CellTiter-Glo® viability assay following six days of incubation with the ADCs and control treatments. Figure 4. Representative dose-response curves (IC₅₀). A) HER2-positive PDXO model comparing exatecan (payload) and trastuzumab deruxtecan (T-DXd) and B) control naked antibody trastuzumab and isotype-payload conjugate IgG-DXd. Across HER2-positive breast cancer PDXO models and control cell lines, ADCs demonstrated greater efficacy and specificity compared with naked antibody and isotype-payload controls (Figure 4, and data not shown). Full datasets across additional PDXO models are presented in the accompanying AACR 2025 poster. These results highlight the utility of PDXO models derived from Champions’ CTG-3D platform for evaluating next-generation ADCs and discriminating between payload-driven cytotoxicity, nonspecific conjugate effects, and HER2-targeted ADC activity. To further quantify differential treatment responses, normalized Area Under the Curve (∆AUC) values were calculated to capture response differences between isotype controls and ADCs across the full concentration range. HER2-positive breast cancer cell lines and PDXO models exhibited higher ∆AUC values (approximately 30–50%, data not shown), indicating stronger ADC activity relative to controls. In contrast, HER2-negative models showed substantially lower ∆AUC values (~20%), consistent with reduced ADC activity in the absence of target expression (Figure 5). Figure 5. Normalized ∆AUC. The response difference between isotypes and ADCs is calculated as: [AUC of Isotype] – [AUC of ADC] / [AUC of Isotype ] x 100% Collectively, these findings demonstrate that breast cancer PDXO models generated from Champions’ CTG-3D platform reproduce expected response patterns to FDA-approved HER2-targeted ADCs with known pharmacological profiles, supporting their utility as a predictive ex vivo platform for ADC development. Key Takeaways Champions’ CTG-3D platform is a biologically relevant ex vivo 3D platform that can support critical preclinical decisions, including payload comparison and mechanism-based screening, enabling ADC candidates to be screened and prioritized before more expensive in vivo evaluation and clinical development is undertaken. For drug developers, this translates into greater confidence in candidate selection, improved translational relevance of preclinical data, and reduced risk of late-stage attrition of potentially viable candidates. By enabling better-informed decisions earlier in the development process, the CTG-3D platform helps accelerate the advancement of safer and more effective ADC-based therapies. To explore the full datasets in more detail, download the full poster presented at AACR 2025 here. References Joubert, Nicolas, Alain Beck, Charles Dumontet, and Caroline Denevault-Sabourin. 2020. “Antibody-Drug Conjugates: The Last Decade.” Pharmaceuticals (Basel, Switzerland) 13 (9). https://doi.org/10.3390/ph13090245.
Evaluating a Novel GCN2 Inhibitor for Acute Myeloid Leukemia (AML) Treatment Using Patient-Derived Models
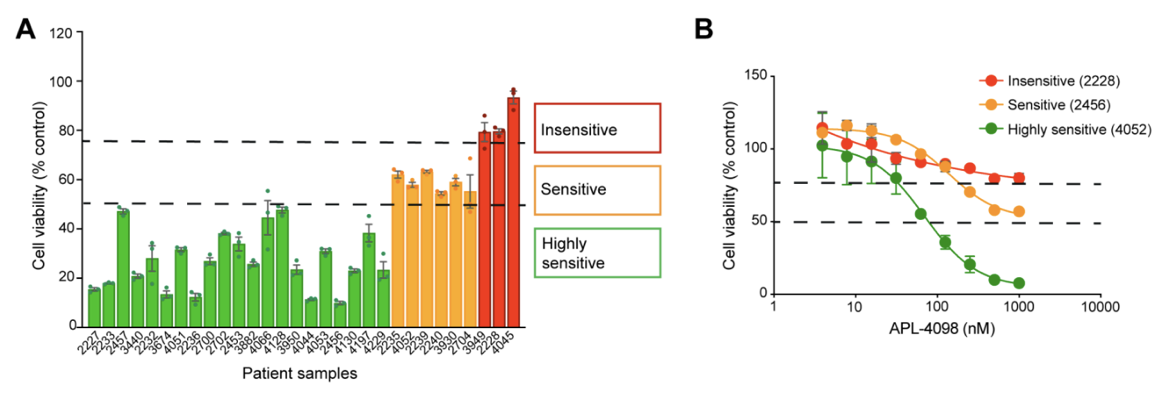
The AML Therapeutic Landscape Relapse remains one of the greatest challenges in treating Acute Myeloid Leukemia (AML). While targeted therapies have improved outcomes for some patients, high relapse rates and limited durable treatment options mean that many patients still face poor prognoses. One pathway gaining attention is the Integrated Stress Response (ISR), a highly conserved protective signaling network that helps cells adapt to nutrient deprivation, oxidative stress, and other metabolic stressors. Many tumor types, including AML, depend heavily on this stress-response system to survive and proliferate. Within this pathway, the kinase GCN2 (General Control Nonderepressible 2) serves as a key sensor of amino acid deprivation and metabolic stress, helping cancer cells survive under unfavorable conditions where lack of exogenous nutrients may disturb intracellular homeostatic balance. Disrupting this survival mechanism has emerged as a promising strategy to selectively target cancer cells and improve treatment outcomes. A recent study explored a novel and selective inhibitor of the stress-response kinase GCN2, APL-4098, as a potential therapeutic approach for AML1. Ex Vivo Evaluation of APL-4098 in Patient-Derived AML Cells Román-Trufero et al. first characterized the anti-leukemic activity of APL-4098 in ex vivo primary patient AML samples using Champions Oncology’s AML VitroScreen platform. A cohort of 30 patient-derived samples was treated with increasing concentrations of the compound and cell viability was measured using a luminescence-based viability readout to determine how effectively the drug inhibited leukemia cell growth. From these results, dose–response curves and IC50 values were generated to quantify drug potency across the cohort. The results revealed strong inhibitory activity across the patient cohort with 70% of samples (21/30) showing a ≥50% reduction in cell viability, indicating high sensitivity to the GCN2 inhibitor. An additional 20% of samples demonstrated some sensitivity, with a 25–50% reduction in viability. Finally, 10% of the samples showed less than 25% decrease in viability and were categorized as insensitive (Figure 1). Figure 1. APL-4098 cytotoxic activity ex vivo. (A) Effect of 500 nM APL-4098 on the viability of patient-derived AML samples after 72h treatment. Values for each specimen are expressed relative to each sample’s matched vehicle-treated control (normalized to 100%) and presented as mean ± SEM, n=3. (B) Selected APL-4098 dose-response curves of highly sensitive (green), sensitive (orange), and insensitive (red) patient-derived samples. Notably, responses were observed across diverse genomic backgrounds, with no clear correlation between common AML mutations and sensitivity. Cells from pre-treated or relapsed patients also responded, suggesting that GCN2 inhibition may be effective even in difficult-to-treat AML. This ex vivo screening step provided critical insight into which patient samples were most sensitive, laying the groundwork for subsequent in vivo evaluation in patient-derived xenograft (PDX) models. In Vivo Evaluation of APL-4098 in AML Patient-Derived Xenografts Following ex vivo screening, the anti-leukemic activity of APL-4098 was further assessed in Champions’ patient-derived xenograft (PDX) AML models. Human AML cells from a patient sample that showed ex vivo sensitivity to APL-4098 were implanted into immunodeficient NOG mice via tail vein injection, allowing leukemia to engraft in the bone marrow. Engraftment of human AML cells in the bone marrow was monitored in surrogate animals using flow cytometry, and once engraftment reached ~20% human CD45+ cells, mice were randomized into four treatment groups: Vehicle control (DMSO) APL-4098 alone (15 mg/kg once daily) Venetoclax alone (100 mg/kg once daily), as a standard-of-care comparator APL-4098 + venetoclax combination Treatments were administered daily for 19 days, and leukemia burden was evaluated by analyzing multiple AML subpopulations, including blasts, progenitors, and leukemia stem cells (LSCs), using multicolor flow cytometry (Figure 2). This allowed researchers to evaluate both single-agent activity and potential synergy with venetoclax in a clinically relevant AML model. Figure 2. Effect of APL-4098, Venetoclax and the combination of both in vivo in an AML PDX model. Bone marrow from AML-engrafted animals was assessed for (A) the percentage of viable human CD45+. (B) the number of viable blasts (CD34+/CD33+/-). (C) the number of viable AML progenitors (CD34+/CD38+) (D) the number of viable LCSs (CD34+/CD38-). In all graphs, data shown as mean ± SEM, statistical significance determined by one-way ANOVA. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. DMSO, n=6; APL-4098, n=7; Venetoclax, n=8, APL-4098 + Venetoclax, n=7. The results showed APL-4098 as a monotherapy had little to no effect on bulk leukemic burden, but preferentially targeted and reduced the LSC-enriched compartment, a niche generally considered primarily responsible for relapse initiation after bulk eradication of leukemic blasts2 Importantly, the combination of APL-4098 and Venetoclax, the standard of care BCL-2 inhibitor in AML patients, produced a pronounced synergistic response, achieving a 97% to 99% reduction across all leukemia subpopulations analysed. Mechanistic studies, including RNA sequencing and metabolic analyses, suggested that APL-4098 exerts its anti-leukemic activity by suppressing mitochondrial respiration, triggering the mitochondrial unfolded protein response, and inducing metabolic stress in AML cells. Together, the researchers demonstrated APL-4098 effectively induces cytotoxicity both ex vivo and in vivo, with preferential effect against the LSC subpopulation, a key driver of patient relapse. While APL-4098 shows potential as a monotherapy agent in the preclinical setting, the potent synergistic interaction between APL-4098 and Venetoclax highlights a potential combination therapy for AML that merits further non-clinical and clinical exploration. This study demonstrates the utility of Champions Oncology’s patient-derived platforms for preclinical and translational AML research, providing a framework to evaluate drug efficacy and explore new therapeutic strategies. To learn more about this study, download the full publication here Explore the Champions Oncology website to discover more about our hematological cancer models and testing capabilities References Román-Trufero M, Whitlock G, Seydoux C, et al. A novel potent and selective GCN2 inhibitor, APL-4098, has anti-leukemic activity through dysregulation of mitochondrial function. Clin Cancer Res. 2026. Hansen Q, Bachas C, Smit L, Cloos J. Characteristics of leukemic stem cells in acute leukemia and potential targeted therapies for their specific eradication. Cancer Drug Resist. 2022.
Not All Tumors Look Alike: Using Surfaceome Diversity to Enrich Responders
The case for measuring the surface directly Biologics and antibody drug conjugates operate at the cell's surface, yet many programs still guide patient selection using RNA expression or whole cell protein abundance. That practice can misclassify candidates because transcripts and total protein often do not correlate, due to post-transcriptional and post-translational mechanisms. Moreover, cellular localization and receptor density can’t be quantified by using whole cell bulk proteomics, therefore not guaranteeing that a receptor is exposed on the exterior membrane above drug relevant thresholds. In 2024, investigators profiled the surfaceome of 100 genetically diverse, primary human AML specimens and resolved antigen patterns on primitive and stem like cells with limited expression in essential normal tissues. The work demonstrated that surface level heterogeneity is real, clinically meaningful, and different from what one would infer from bulk RNA or whole cell proteomics alone. Cell+1 Calls to better map the surfaceome are growing. A 2024 Cancer Discovery commentary noted that only a few dozen cell surface targets currently anchor FDA or EMA approved therapies, despite the surface being the most accessible compartment for antibodies, CAR T cells, and radiopharmaceuticals. The authors argued for intensified efforts to chart the universe of surface proteins across cancers, which would accelerate target discovery and improve translational relevance for modalities that require true surface accessibility. AACR Journals+1 Solving the Surfaceome Problem Why a surface resolved view changes development plans Modern reviews describe how advances in mass spectrometry, fractionation, and enrichment are making it feasible to survey the cancer surfaceome at scale. These approaches improve the identification of drug accessible proteins that whole cell proteomics can miss, because abundant intracellular proteins dominate unfractionated measurements and mask low abundance, surface localized receptors. The reviews also outline practical ways to mitigate technical challenges such as low copy number, hydrophobic domains, and contamination by intracellular compartments, for example through careful membrane preparation, parallel intracellular and whole cell fractions, and stringent enrichment and quality control thresholds. The program level implication is straightforward. When a therapy acts at the surface, the biomarker strategy should resolve surface localization directly rather than infer it from RNA or whole cell protein alone. PubMed+1 A second implication concerns internalization and trafficking. For ADCs and some bispecifics, internalization kinetics and routing to lysosomes affect payload delivery and potency. Surface resolved workflows can be coupled to orthogonal assays, for example flow cytometry with ligand induced internalization or live cell imaging, to determine whether a receptor is not only present at the surface but also behaves in a way that supports the intended mechanism. This behavioral dimension rarely appears in transcript based selection but influences both efficacy and safety. From heterogeneity to practical enrichment Turning surface heterogeneity into clinical signal requires a sequence of disciplined steps. First, measure what matters. Use fractionated surface proteomics or validated surface specific immunohistochemistry and flow assays that distinguish the exterior membrane from total abundance. Second, define actionable thresholds that tie expression to benefit and that can be reproduced across sites. Educational content from the ASCO Educational Book emphasizes that quantitative cutoffs and standardized assays are central to patient selection for antibody drug conjugates, because target accessibility and abundance determine benefit, and because inconsistent thresholds erode the interpretability of early phase studies. Third, map prevalence by histology and by clinically relevant subgroups, since an attractive target with low prevalence may not support enrollment or may require a targeted site strategy. When these elements are present, enrichment reflects how the drug actually works and not a proxy signal. ASCO Publications+1 Surface level diversity can also sharpen indication strategy. If a receptor is highly enriched at the surface in a subset of colorectal cancer but not in pancreatic cancer, a program can prioritize the former for first in human evaluation, even if RNA levels are similar in both. This is particularly relevant where surface and whole cell abundances diverge. The AML study noted above showed precisely this phenomenon, where antigen exposure on stem like compartments could be quantified and connected to therapeutic concepts that require surface binding. Cell Selectivity and safety as design constraints Surface heterogeneity intersects with safety because many candidate antigens have some expression in normal tissues. A 2024 review in Trends in Pharmacological Sciences surveyed strategies that increase antibody selectivity in oncology, including superselectivity through avidity and multivalency, conditional or pH sensitive binding, dual targeting that requires co expression to achieve high affinity, and engineering designs that leverage tissue context. These concepts translate directly into earlier program decisions. Targets that can be paired with selective engineering approaches should score higher than those that would require unrealistic discrimination from a conventional binder. A transparent scoring framework, for example one that penalizes normal tissue expression in essential organs and rewards tumor specific co expression patterns, makes those choices auditable during governance reviews. Cell+1 The safety argument is not theoretical. ADCs can cause off tumor toxicities when a payload is delivered to normal tissues that express the target at modest levels. A biomarker plan that quantifies true surface exposure in disease and screens for surface exposure in a curated panel of normal tissues improves the chance of a workable therapeutic window. Reviews that focus on ADC biomarkers, together with trial design guidance, point in the same direction, namely that selection should be quantitative and assayable, not an exploratory cutpoint chosen after the fact. ASCO Publications Concrete examples of surfaceome driven discovery Evidence that surface resolved discovery translates into programs is accumulating. In late 2024, a Cancer Cell study used an integrative proteogenomic surfaceome approach to credential DLK1 as an immunotherapeutic target in neuroblastoma. The authors combined mass spectrometry based surface profiling with genomic and transcriptomic context to demonstrate surface exposure, tumor specificity, and functional relevance, then moved to validation experiments that supported drug development. This work illustrates how surface level datasets, anchored in clinical material, can identify and qualify targets that a transcript only screen might underrate. PMC+3Cell+3PubMed+3 Surfaceome maps are also becoming more accessible, which will help teams generalize findings across institutions. The AML dataset is public in GEO, enabling independent inspection of QC criteria, antigen lists, and statistical methods. Commentary from Cancer Discovery underscores the systemic opportunity, arguing that better cartography of the surfaceome across cancers is likely to grow the small set of currently actionable surface targets. Together, public data and field level commentary support a move from opportunistic target picking toward systematic, population informed discovery. NCBI+1 TALK Decoding the Cell Surface to Accelerate Discovery Program design, trial execution, and measurement Programs that embrace surface resolved selection can operationalize three habits that improve downstream signal. First, connect discovery assays to clinical screening early. If discovery uses a fractionated surface proteomics threshold, define the clinical assay that will mirror that readout, for example an IHC score or a validated flow protocol, and harmonize cutoffs before first patient in. Second, pre specify enrichment rules in the protocol. Enrolling only the top percentiles of surface expressors may seem restrictive, but it increases the chance of a pharmacodynamic signal that validates the mechanism and informs dose expansion. Third, measure what happens when selection criteria are varied in sensitivity analyses, and share these details in publications and regulatory interactions. This transparency builds cumulative knowledge that benefits the field and informs the next iteration of thresholds. There is also value in linking surface metrics to pharmacology. If a surface antigen is abundant but shows slow internalization, a payload with a bystander effect may be preferable to a payload that requires rapid lysosomal routing. If surface expression is heterogeneous at the lesion level, a radionuclide therapy that can exploit crossfire may offer advantages over a conventional ADC. These are not general rules, they are examples of how a surface resolved view can shape the choice of modality and payload in a way that reflects the physical constraints of the target. Where Champions Oncology contributes Champions Oncology generates surface resolved datasets from low passage, clinically relevant tumor models and integrates them with deep multi omic profiles and in vivo pharmacology. This enables teams to quantify true surface positivity, to set and test actionable thresholds, and to understand prevalence by indication before committing to costly trials. The same datasets support orthogonal validation and method standardization so that discovery assays translate into clinical screening. The approach is designed to be non promotional and data first, the objective is to clarify risk and to help programs make better decisions earlier.
Beyond Single Omic Biomarkers: How Proteogenomic ML Reveals Therapy Vulnerabilities
Why a functional view changes predictions Genomics and transcriptomics remain foundational for precision oncology, but they do not fully represent the functional state that determines how tumors respond to therapy. Proteins and phosphoproteins capture activity at the level where drugs actually engage, for example receptor density and localization, complex assembly, and pathway signaling. That distinction is not academic. In a 2024 pan-cancer analysis from the Clinical Proteomic Tumor Analysis Consortium (CPTAC), investigators integrated proteogenomic data from 1,043 patients across 10 tumor types, surveyed 2,863 druggable proteins, and quantified biological factors that weaken mRNA to protein correlation, making the case for models that learn directly from protein and phosphoprotein context rather than inferring from transcripts alone. Cell From data to models that travel The practical bottleneck has been access to harmonized, well-annotated cohorts that support training, testing, and independent validation. In August 2023, the National Cancer Institute announced a standardized pan-cancer proteogenomic dataset that aligns genomics, proteomics, imaging, and clinical data for more than 1,000 tumors across 10 cancer types, explicitly to enable reproducible discovery and model benchmarking. The Proteomic Data Commons (PDC) now serves these resources in a way that supports programmatic access and cross-study comparisons, a requirement if machine-learning outputs are going to generalize beyond a single study. National Cancer Institute+1 Solving the Surfaceome Problem What the evidence shows when proteins are included Two Cell papers from 2024 illustrate why adding protein-level information changes conclusions. An immune-landscape analysis derived distinct immune subtypes by integrating genomic, epigenomic, transcriptomic, and proteomic features and connected oncogenic drivers to downstream protein states that influence immune surveillance and evasion. A companion pan-cancer study expanded the landscape of therapeutic opportunities by evaluating thousands of druggable proteins across tissues and documenting where mRNA is a poor proxy for protein, especially in pathways relevant to therapy response. Together, they show that multi-omic modeling, including protein and phosphoprotein features, improves biological interpretability and exposes actionable biology that single-omic approaches overlook. Cell+1 A broader signal from the field The trend is not confined to CPTAC. The Pan-Cancer Proteome Atlas (TPCPA), published in Cancer Cell in 2025, quantified 9,670 proteins across 999 primary tumors representing 22 cancer types using DIA-MS. The atlas offers a tissue-based substrate for target nomination, biomarker discovery, and external validation, and has been highlighted in the trade press for its global availability and immediate relevance to oncology research. Such atlases are valuable because they capture proteomic variability directly in clinical material, not only in cell lines, providing realistic distributions for features that ML models attempt to learn. Cell+2PubMed+2 Why proteogenomic ML improves prediction Integrating proteins and phosphoproteins adds information that is both mechanistic and measurable. First, pathway activity is reflected in phosphorylation states, which function as on–off or rheostat-like controls for signaling. Second, receptor exposure and complex formation at the protein level determine whether a therapy can bind or disrupt a process. Third, protein degradation and post-translational regulation often decouple mRNA abundance from target availability, which explains why transcript-only biomarkers can fail at the bedside. When these features are engineered into models, performance gains are not just numeric; they tend to be more interpretable, mapping to drug-actionable pathways and receptors that clinicians recognize. The 2024 CPTAC studies provide concrete examples. Immune subtypes defined by proteogenomic features correlate with differences in antigen presentation, cytokine signaling, and interferon responses, features with obvious translational implications. The survey of druggable proteins shows wide variation in abundance and localization across tumors and details the contexts where transcript and protein diverge, arguing for protein-aware rules when nominating targets or stratifying patients. Cell+1 What good practice looks like in model building There is a growing consensus on practical guardrails. Independent validation across cohorts is essential to avoid overfitting, and the infrastructure now exists to support that step through the PDC and related CPTAC resources. Feature construction should prioritize pathway-level signals that aggregate individual phospho-sites into kinase or pathway activity because these are more stable across cohorts and easier to interpret for clinical decision making. Finally, clinically annotated samples, including treatment history and outcomes, are indispensable if models are expected to inform responder enrichment and mechanism-of-resistance hypotheses rather than only classify molecular subtypes. National Cancer Institute Translational payoffs, with appropriate caution When executed with these guardrails, proteogenomic ML offers tangible benefits. Programs can generate earlier responder and non-responder hypotheses and test them prospectively in preclinical systems before committing costly clinical designs. Resistance pathways inferred from phospho-proteomic features can motivate combination strategies, for example pairing an antibody-drug conjugate with a kinase inhibitor when signaling indicates a plausible escape route. Educational content from ASCO has emphasized the centrality of quantitative thresholds and validated assays for patient selection, particularly for ADCs where surface accessibility and abundance determine benefit. The lesson is consistent across modalities. Predictive features must be connected to assays that can be deployed consistently in trials, and thresholds should be defined in a way that anticipates real-world variability. PubMed Where limitations still matter Several limitations deserve explicit mention. Proteomic and phospho-proteomic data remain technically variable across platforms and laboratories. Although CPTAC and PDC mitigate this through standardization, modelers should evaluate batch effects and apply normalization strategies suited to proteomic data. Coverage of kinase–substrate relationships and post-translational networks is incomplete, which constrains inference. Tumor heterogeneity adds another layer, particularly when bulk tissue averages mask subclonal or microenvironmental signals. These caveats do not negate the value of proteogenomic ML, but they do argue for conservative claims, orthogonal validation, and a bias toward features that can be measured reproducibly in clinical settings. TALK Decoding the Cell Surface to Accelerate Discovery Implications for trial design and portfolio focus The immediate implication is a more disciplined approach to enrichment. If protein-level features identify a subgroup with plausible sensitivity, early designs can incorporate eligibility criteria and stratification based on validated assays rather than exploratory cutpoints. Conversely, if pathway-level features suggest multiple escape routes, it may be more efficient to prioritize combinations earlier instead of iterating single-agent studies. At a portfolio level, proteogenomic evidence can help prioritize programs with a mechanistic rationale supported by functional data, not only by mutation prevalence or gene expression. How Champions Oncology contributes Champions Oncology builds models on tumor-derived systems that preserve patient biology and heterogeneity. Our datasets link genomics and transcriptomics with proteomics, phospho-proteomics, and cell surface proteomics, and they are annotated with pharmacologic phenotypes. This combination supports models that tie features to functional biology and drug accessibility, making it possible to move from correlation to causal relation and from causal relation to druggable targets.

Feature selection in RNA-seq and proteomics with MADVAR in R
High dimensional omics datasets often include many features that contribute little to downstream analysis. This can blur structure in unsupervised tasks, slow computation, and complicate model training. The MADVAR study introduces two simple, data driven procedures that set feature selection thresholds from the distribution of the data itself, rather than relying on fixed heuristics. The first procedure, madvar, computes a variance cutoff using the median plus a multiple of the median absolute deviation. The second, intersect Distributions, fits a two component Gaussian mixture to the variance or another continuous score, and uses the intersection point between components as the cutoff. Both methods are implemented in an R package and were evaluated across public datasets that include TCGA gene expression, GTEx proteomics, and CPTAC phosphoproteomics. The paper reports improvements in unsupervised clustering quality and competitive supervised performance with fewer features, while keeping runtime and memory use modest. What the Paper Tested The benchmarking examined unsupervised structure and supervised classification. For unsupervised analysis, the authors applied filtering and then assessed cluster quality with connectivity, the Dunn index, and the Biological Homogeneity Index. Across datasets, the variance based approaches produced tighter or more homogeneous clusters on these metrics. For supervised analysis, they trained random forest models with repeated runs. Both approaches produced low out of bag error rates. Retaining more features sometimes improved accuracy, but MADVAR often matched the mixture based approach while selecting fewer features. The paper also documents practical defaults, such as Ward.D for hierarchical clustering with Euclidean distance, and explains how to pass either a raw matrix or a precomputed variance vector into the functions. Source code and documentation are available on GitHub. When These Methods are a Good Fit These procedures are particularly well suited for rapid, large-scale preprocessing, when analyses require a quick, efficient, and transparent approach to feature selection prior to dimensionality reduction, clustering, or model fitting. They also integrate naturally into interpretability-oriented pipelines, since thresholds based on medians or mixture-model intersections are simple to explain and justify to collaborators. Because the logic operates on continuous feature scores, the same framework can be applied seamlessly to any quantitative data type that can be summarized by variability Things to keep in mind. Variance is a proxy for informativeness, not a guarantee. Low variance does not always mean a feature is uninformative. Some biomarkers remain stable yet become predictive in combination with others. If domain knowledge indicates a feature should be preserved, the package allows must keep lists. The mixture based method assumes the variance distribution resembles a two-component mixture. If the fit is poor, the intersection may not be meaningful, so density plots are worth inspecting before adopting the cutoff. Downstream metric choice also matters. Gains in the Biological Homogeneity Index or Dunn index describe cluster characteristics, which may not translate to improvements on other endpoints such as survival modeling or dose response prediction. Finally, supervised performance can depend on class imbalance and sample size. If your data are skewed, tune the learner and validate with a scheme that reflects your use case. Read the Full Paper How to Apply, a Straightforward Workflow A practical workflow starts by exploring the variance distribution. Plot the density (using the madvar flag `plot_density = TRUE`), confirm whether there is a near zero peak, and decide whether a MAD based threshold or a two-component mixture are appropriate. Set a conservative first pass using the default MAD multiplier (`mads = 2`) and adjust if another stringency level is preferred, or, if you prefer the mixture approach, verify the intersection visually before you commit to the cutoff. Preserve domain critical features by whitelisting known markers or controls that should not be dropped. Re run the planned clustering or model fitting, compare structure and error rates before and after filtering, and record any change in feature counts and compute time so the impact is transparent to collaborators. Reproducibility and availability The R implementation and documentation are available on GitHub, as referenced in the paper. The evaluations draw on TCGA gene expression, GTEx proteomics, and CPTAC phosphoproteomics, with figures that show density plots, clustering metrics, and classification results. The article appears as an Application Note in Bioinformatics Advances and is available for open access. Summary MADVAR provides two transparent, variance-based rules for feature selection, enabling the removal of near invariant features from large omics matrices. In the reported benchmarks, these procedures improved or maintained clustering quality and supervised accuracy while substantially reducing feature counts and computational load. The approach is easy to inspect, easy to explain, and simple to integrate into existing R workflows. As with any filter, final value depends on the analysis goal, so it is worth validating its effects on the endpoints that matter for your study. Silberberg G. “MADVAR, a lightweight, data driven tool for automated feature selection in omics data.” Bioinformatics Advances 2025, vbaf211. doi, 10.1093/bioadv/vbaf211. Explore our Data Ecosystem

Choosing the Right CDX Models: Speed, Scientific Value, and Translational Relevance
In oncology drug development, cell line-derived xenograft (CDX) models remain one of the cornerstone in vivo tools for evaluating new therapies in the preclinical ecosystem. They offer a balance of biological relevance, reproducibility, and speed that makes them ideal for early phase hypothesis testing. Nevertheless, while CDX models are indispensable for initial target screening and validation, they also have well-understood limitations that can upset downstream progress if not properly accounted for. As drug developers increasingly rely on CDX-based systems to screen and prioritize compounds before moving into clinically relevant, albeit more expensive, patient-derived xenografts (PDX), the need for appropriate model selection has become critical. The right CDX program can deliver early translational clarity and strategic focus, while the wrong one can generate noise that obscures the true efficacy of an otherwise promising therapeutic, potentially derailing its continued development. Balancing Speed and Fidelity CDX studies are often designed for speed. They enable large-scale screening of novel agents, as both mono- and combination therapies, and dosing regimens in a fraction of the time and cost required for PDX studies. This makes them especially valuable at the earliest stages of decision-making, where timelines are incredibly compressed and attrition risk is high. But that same speed exacts a cost. Many CDX models originate from long established cell lines that have been maintained in culture for decades. Over time, these lines lose virtually all the critical genomic and cellular heterogeneity, stromal interactions, and microenvironmental complexity characteristic of a bona fide tumor. Whilst they grow predictably, the biological features they retain may no longer reflect that of tumors seen in clinical patients. However, the key is not to abandon CDX models, but to recognize where they fit in the development pipeline and to acknowledge and mitigate the limitations they have. Used prudently, CDX models are an efficient and scientifically powerful system to rank compounds, explore mechanisms, and refine hypotheses before moving into PDX for deeper translational validation. Defining a High Value CDX Model The quality of a CDX model is defined by its biological fidelity and characterization depth. Models derived from contemporary, clinically annotated cell lines are more likely to capture the genomic and phenotypic diversity relevant to modern oncology therapeutics. When models reflect the intrinsic complexity of current patient populations, such as the plethora of KRAS G12 mutations or the legion mechanisms by which EGFR becomes hyperactivated, they offer drug developers meaningful results and mechanism-linked insights that can inform clinical strategy. Biological and “omic” characterization matters as much, if not more, than cellular/tissue origin in CDX models, particularly as clinical oncology continues to diverge from tissue-based therapy to therapeutic roadmaps grounded in the molecular features of each patient tumor. A high value CDX model is supported by multi-omic profiling that includes genomic, transcriptomic, and proteomic annotation. This data allows researchers to interpret observed drug effects through the lens of pathway activation, resistance mechanisms, and biomarker expression. In contrast, models lacking such characterization risk generating results that are descriptive rather than explanatory. Drive Proof of Concept and Target Validation with Champions’ CDX Models Mechanistic Clarity Through Multi-Omic Integration The rise of precision oncology has shown that pharmacology and data science are inextricably linked. Researchers now expect and rely on preclinical models to yield mechanistic understanding and insight, not just tumor regression rates. Integrating omic datasets into CDX studies transforms them from mere screening tools into translational resources capable of generating biological comprehension and preclinical direction. For instance, RNA sequencing of treated and untreated xenografts can reveal transcriptional signatures underpinning positive response outcomes, potentially allowing clinical partitioning of patients likely to receive the most benefit from a therapy. As another example, phosphoproteomic profiling can identify compensatory signaling cascades mediating adaptive drug resistance, permitting de novo combination therapies to be trialed to preempt such resistance before it evolves. This approach enables drug developers to anticipate how tumors might evade inhibition, long before clinical exposure. Moreover, omic integration provides a framework for cross platform alignment. Data from CDX models can be mapped against PDX datasets, public repositories, or patient trial cohorts, accelerating the feedback loop between preclinical findings and clinical validation. Modeling Resistance and Tumor Evolution One of the most powerful applications of CDX technologies is in modeling acquired resistance. By exposing tumor-bearing mice to sustained drug pressure, scientists can select for resistant clones that mimic clinical relapse. Comparative molecular profiling between parental and resistant CDX lines may illuminate the pathways that drive therapeutic escape, whether through secondary genomic changes, activation of bypass signaling cascades, or metabolic rewiring. This approach supports the rational design of next-generation inhibitors or combination strategies aimed at delaying or overcoming resistance. It also informs biomarker development by revealing the early molecular changes that forecast reduced drug sensitivity, enabling the design of clinical trials with built-in resistance monitoring. Bridging the Gap to PDX Despite their wide-ranging utility and flexibility as experimental tools, CDX models are at best a starting technology in the developmental pipeline. Translationally-minded organizations deploy CDX models as a filter for promising candidates in an experimental continuum that leads naturally into PDX. PDX models, established directly from patient tumors, preserve the architecture, stromal components, and molecular heterogeneity of the original cancer. They capture biological features that CDXs inherently lack, including contextualized immune responses in humanized systems, evolution and expansion of tumor sub-clones, and the influence of the microenvironment on cancer progression. For these reasons, PDX validation remains an important next step, if not a critical one, once a compound demonstrates clear activity in CDX. The most efficient development pipelines are those where CDX and PDX models are in biological alignment, where both originate from well characterized sources and overlap genomically and/or phenotypically. In this context, compound evaluation flows smoothly from screening to translational evaluation. A consistent molecular linkage between model systems strengthens the predictive bridge and ensures that early results translate more faithfully into clinical outcomes. Speed Without Sacrificing Relevance The enduring appeal of CDX model systems lies in the speed with which large quantities of data can be generated to reinforce or oppose development of individual drug compounds, or indeed entire drug programs. Studies can be initiated quickly, with timelines to results measured in weeks rather than months. Moreover, CDX models can support the simultaneous exploration of multiple therapeutic hypotheses. Where delays mean patients remain beset with therapeutic inadequacies and can cost developers millions in lost opportunity, this speed is a major competitive advantage. But whilst speed is a necessary component of drug development, it is insufficient for success. The most effective drug pipelines are designed with translational intent incorporated into CDX programs from the outset. CDX model selection is based on mechanistic alignment derived from deep omics comprehensive characterization, and the clinical transition to PDX model systems is a crucial element of the sequence rather than an unintegrated effort. This approach ensures CDX models are deployed as value-enhancing study tools,leveraging the efficiency of CDXs to inform smarter, faster progression into the PDX models best approximating patient clinical features and responses. Designing a Modern CDX Strategy For biotechnology and pharmaceutical teams, a modern CDX strategy balances three principles:speed, depth, and connectivity. Speed means using CDX models to quickly triage candidate molecules, confirm on-target effects, and eliminate ineffective compounds prior to larger resource investment. Depth refers to multi-layered omics characterization to uncover mechanistic drivers of response or resistance. And finally, connectivity means designing CDX studies with the downstream transition to PDX models in mind, ensuring molecular alignment and continuity between the different systems. When these principles are applied judiciously, CDX models become a strategic asset that accelerates development timelines without compromising scientific integrity. The Future of Translational Modeling As oncology continues to evolve toward precision medicine, the most impactful preclinical programs will be those that connect fast data generation with deep omics characterization, using rapid CDX screening to guide more complex, patient-relevant studies. Emerging approaches such as multi-omic analytics, AI-driven model selection, and ex vivo/organoid validation are expanding how CDX data can inform clinical decision-making. Taken in concert, these all suggest a future where the value of a model is defined not only by how fast it can be employed to produce data, but also by how that same data can be used to map the necessary downstream steps to ensure successful drug development and patient application. Choosing the Right Partner In an increasingly competitive preclinical landscape, the distinction between a vendor and a scientific partner has never been clearer. The most valuable CDX programs combine biological relevance, data transparency, and translational foresight. When evaluating potential collaborators, sponsors should ask not only what models are available but how those models were developed, characterized, and validated. The answers will reveal whether a platform can deliver more than results, whether it can deliver understanding. Cell Line Select Tool A smarter way to explore Champions’ cell line models.
Modeling next generation AR pathway inhibitors in prostate cancer
When Inhibition Isn’t Enough: How Dual-Mechanism AR Degraders Are Redefining Resistance Modeling For over a decade, androgen receptor (AR) pathway inhibitors such as enzalutamide and abiraterone have formed the foundation of treatment for advanced prostate cancer. Yet the same story continues to unfold patients initially respond, then relapse. Despite continued suppression of androgen signaling, tumors adapt reactivating the AR axis through overexpression, gene amplification, or ligand-binding domain mutations. What follows is a return of disease activity that current inhibitors can no longer control. This persistent pattern highlights a central truth in oncology drug development: inhibition alone is not enough. Over time, cells find ways to reengage the same signaling pathways that were once silenced. A new class of therapies is changing that. Bristol Myers Squibb recently published a landmark study in Clinical Cancer Research describing BMS-986365, a first-in-class dual AR degrader and antagonist. By combining proteasomal degradation with receptor antagonism, BMS-986365 achieves a more profound and sustained blockade of AR activity than traditional inhibitors. The drug not only shuts down signaling but also removes the receptor protein responsible for resistance and relapse. Learn more about our Prostate Models Champions Oncology’s patient-derived xenograft (PDX) models CTG-2440 and CTG-2441 were instrumental in this discovery. These models were derived from the same patient before and after abiraterone therapy, creating a unique, clinically matched system for studying adaptive resistance. In both models, BMS-986365 was tested alongside enzalutamide to evaluate its impact on AR signaling in tumors that had already progressed on prior therapy. While the model developed before the patient received abiraterone showed modest increased sensitivity to BMS-986365 compared to Enzalutamide, BMS-986365 outperformed Enzalutamide when administered to the model developed after the patient progressed under abiraterone. Both drugs increased AR mRNA expression in treated tumors, q a typical feedback response to pathway inhibition but only BMS-986365, by virtue of its degrader activity, maintained low AR protein levels and continued to suppress AR target gene activity. This finding underscores the importance of degradation: while transcriptional upregulation persisted, the protein was degraded before it could restore signaling. The biological feedback loop driving adaptive resistance was disrupted during treatment. These insights speak to a broader principle in translational research: resistance is dynamic, not static. It doesn’t exist as a single genetic event, but as a continuum of cellular responses that evolve under therapeutic pressure. Capturing this dynamic behavior requires preclinical models that replicate the complexity of human disease including treatment history, adaptive feedback mechanisms, and the heterogeneous signaling environments of late-line tumors. For drug developers, this means that testing next-generation degraders and dual-mechanism agents cannot rely on traditional cell lines or simplified, outdated in vivo models. The field now demands functional, clinically grounded resistance models that measure more than endpoint response they must reveal how and why tumors adapt, and how new modalities can overcome that adaptation. Champions Oncology’s portfolio of pretreated and resistance-matched PDX models was built precisely for this purpose. By recreating clinical resistance within the same biological context in which it arises, these models provide a high-fidelity platform to evaluate degrader pharmacodynamics, durability of response, and combination potential. The BMS-986365 study offers a clear demonstration of their value: real-world resistance biology, translated into preclinical discovery that informed a novel therapeutic strategy. The success of BMS-986365 represents more than a promising drug—it marks a shift in how we define preclinical relevance. By pairing innovative therapeutics with equally advanced resistance models, the field is beginning to close the translational gap that has long limited success in late-stage prostate cancer. Discover Pretreated and Resistance-Matched PDX Models

Reducing Clinical Attrition: Why Stronger Data Needs to be the Starting Point for Oncology R&D
Clinical attrition has been oncology’s oldest problem and, in many ways, still its biggest. The pattern is painfully familiar. A promising therapy emerges with encouraging preclinical data, advances through IND-enabling studies, shows early signals of activity in Phase I, and then fails in Phase II or Phase III. The financial costs of these failures are staggering, billions of dollars are lost globally each year. But the greater cost is measured in time and opportunity, years of development work invested, only to leave patients still waiting for new therapies. Despite decades of innovation, attrition rates in oncology haven’t shifted as much as the industry hoped. Better trial design and precision medicine strategies have helped in some areas, but the fundamental problem remains: the data we use to make early decisions often doesn’t capture the full reality of patient biology. Decoding the Cell Surface to Accelerate Discovery Why attrition remains so stubborn To understand why attrition persists, it’s worth looking at the foundation. Much of oncology R&D still relies on models and datasets that, while powerful, were never meant to carry the full burden of translational decision-making. Genomics is a prime example. Sequencing technologies have revolutionized how we classify tumors and identify potential targets. But tumors are not defined by their mutations alone. Transcriptional programs, proteomic signaling networks, post-translational modifications, and dynamic adaptations under treatment all contribute to how a tumor grows, evades therapy, and eventually resists intervention. A therapeutic strategy built solely on genetic alterations may miss the downstream biology that ultimately determines clinical outcome. Cell lines are another example. They are convenient, reproducible, and cost-effective, which is why they remain a staple of preclinical research. But they lack the heterogeneity and clinical context of patient tumors. They rarely reflect the complexity of pretreated, metastatic disease — exactly the patient populations that new oncology drugs are tested in. When early models don’t reflect the biology of the intended clinical population, it is not surprising that translation breaks down. Even when multi-omic data is available, it is often sparse, fragmented, or drawn from public repositories that were never built for translational research. These datasets may be useful for generating hypotheses, but they are rarely robust enough to support critical go/no-go decisions. And yet, in the absence of better resources, they are often asked to do just that. The gap between data and patients The result of this reliance on incomplete models is a gap between what we believe about a therapy and what happens when it is tested in patients. That gap is where attrition lives. It’s the difference between a drug that looks compelling in preclinical settings and one that can’t demonstrate sufficient efficacy or durability in the clinic. One concrete example comes from RNA and protein data. In acute myeloid leukemia (AML), large-scale analyses have shown that only about 17% of genes have a positive correlation between RNA expression and protein expression. That means if you are relying on transcriptomics alone to predict biology, you’re often looking at signals that don’t translate to the level where drugs actually act. This divergence isn’t unique to AML — it’s a reminder that single-omic views can give an incomplete or even misleading picture of tumor biology. Another example is in resistance biology. In pretreated patient-derived xenografts (PDX), resistance pathways are often “baked in” from the start, reflecting real-world clinical histories. These mechanisms are invisible in naïve cell lines, which haven’t experienced therapy. By working with tumors that already carry resistance features, researchers can anticipate escape mechanisms before they derail late-stage trials. What better data could look like If we accept that the root of the problem lies in the misalignment between early data and patient biology, then the question becomes: what would better data look like? First, it would need to come from models that are closer to the clinic. Patient-derived tumors, especially those from pretreated and metastatic populations, preserve the genetic complexity, phenotypic heterogeneity, and resistance mechanisms that cell lines cannot replicate. Studying these tumors allows us to see not just what cancer looks like in theory, but how it behaves in practice. Second, it would need to move beyond genomics into multi-omic depth. Genes matter, but so do the transcripts they produce, the proteins they encode, the phosphorylation states that regulate those proteins, and the cell surface markers that mediate interactions with the immune system or targeted therapies. Each of these layers adds context. And critically, each reveals discrepancies that can’t be seen in isolation. Take cell surface proteomics as an example. Traditional workflows for mapping the “surfacome” are plagued by noise and misclassification, which can lead to wasted effort on false targets. By capturing both plasma membrane and intracellular fractions, newer approaches provide cleaner enrichment and reduce false positives. The result is surface protein datasets that can actually be used to prioritize antibody, ADC, or CAR-T targets with confidence. That’s not a small improvement — it’s the difference between pursuing targets that work in patients and chasing dead ends. Third, it would need to incorporate functional context. Static descriptions of tumors, no matter how deep, tell us what’s there, but not how the tumor behaves under pressure. Functional assays that perturb tumors directly, whether through gene knockdowns or compound exposure, provide causal insights that correlation alone cannot. They show us how pathways respond, how resistance emerges, and how biology adapts. For example, siRNA knockdown studies in 3D PDX models can reveal dependencies that aren’t obvious from genomics alone. When combined with high-resolution transcriptomic profiling (what we call FunctionalSeq), these experiments identify pathways that are not only present but functionally essential. That’s the kind of information that can distinguish a biomarker from a true therapeutic target. What this means for pharma decision-making For pharma R&D leaders, the implications of this kind of data are significant. Instead of evaluating a candidate on a narrow slice of biology, you can assess it in the context of real patient tumors, profiled across multiple dimensions. You can compare across cohorts, understand potential resistance pathways earlier, and align therapeutic strategies with the biology most likely to be encountered in the clinic. Consider the decision to advance an asset into IND-enabling studies. In many organizations, this call is based primarily on genomic alignment, preliminary efficacy signals, and a limited view of resistance. Adding multi-omic and functional data changes the conversation. It allows teams to say, “Yes, the target is present at the DNA level, but the protein expression isn’t concordant,” or, “The mechanism looks strong in cell lines, but resistance emerges rapidly in pretreated PDX.” These insights don’t just inform science — they directly affect which assets receive investment and how development strategies are shaped. A future with fewer blind spots Attrition will always be a risk in oncology. Biology is unpredictable, and even the most carefully designed program may fail in the clinic. But the scale of today’s attrition, and the cost it imposes does not have to be inevitable. By aligning our early data more closely with patient reality, we can reduce blind spots, strengthen translational confidence, and make smarter decisions about which programs deserve to move forward. For pharma leaders, the payoff is not just fewer late-stage failures. It’s a more rational, efficient, and patient-centered pipeline. And for patients, it’s a better chance that the therapies entering trials are the ones with the greatest likelihood of success. That is the promise of stronger data and the reason it should be the starting point for oncology R&D. This isn’t just data. It’s a foundation for discovery.
Radiopharmaceutical Efficacy Studies in PDX Models: Why Tumor Diversity Matters
As radiopharmaceutical therapies move from proof-of-concept to clinical investment, preclinical efficacy data plays an outsized role in shaping go/no-go decisions. But not all efficacy studies are created equal. Traditional in vivo models often fall short in capturing the biological variability that influences drug performance in patients leading to overly optimistic interpretations and costly setbacks downstream. That’s where patient-derived xenograft (PDX) models provide a distinct advantage. Unlike cell line–based systems, PDX models preserve the molecular, phenotypic, and histological heterogeneity found in human tumors, enabling more realistic evaluations of how radiopharmaceuticals behave across diverse tumor types. In this post, we explore how tumor diversity impacts radiopharmaceutical efficacy readouts, why it matters for translational success, and how the right PDX strategy can strengthen early decisions on compound prioritization, dose optimization, and biomarker alignment. PDX Models + Radiopharmaceuticals = Translational Power The Challenge with Cell Line–Based Tumors in Preclinical Efficacy? Conventional xenograft models, particularly those directly derived from established cell lines (CDX), have been a mainstay of preclinical oncology research. Their reproducibility and ease of use make them convenient, but their biological homogeneity is also their greatest limitation, especially for evaluating targeted therapies such as radiopharmaceuticals. CDX models typically originate from immortalized cell lines propagated in vitro for years. As a result, they display homogeneous antigen expression, clonal architecture, simplified stromal and vascular features, and a lack of microenvironmental complexity. These characteristics often inflate perceived efficacy. Uniform antigen presentation can lead to idealized tumor uptake, while consistent growth kinetics and structure can overstate the durability and magnitude of response. Drugs that appear highly potent in CDX models often underperform when tested against the biological heterogeneity of patient tumors. Because CDX models fail to capture inter-patient and intra-tumoral variability, they offer little insight into how a drug might behave across subsets of patients. That gap matters, since therapeutic index, antigen density, and radiosensitivity all vary widely in the clinic. The “clean” signals that CDX models produce may look promising on paper but can be misleading. For radiopharmaceutical programs, where tumor retention, dose-response, and antigen heterogeneity shape success, more clinically faithful models are essential. How Tumor Diversity Affects Radiopharmaceutical Response Radiopharmaceutical efficacy depends heavily on biological context. Antigen density, vascular accessibility, tumor perfusion, and radiosensitivity all differ not just from patient to patient, but even across different regions of the same tumor. This variability shapes how radiopharmaceuticals behave in vivo. Heterogeneous antigen expression may result in only partial tumor coverage, which reduces therapeutic effect. Vascular differences and interstitial pressure can limit isotope delivery and retention. Radiosensitivity, influenced by DNA repair pathways, hypoxia, and tumor subtype, alters how readily tumors undergo radiation-induced cell death. Models that reflect this complexity are critical for generating data that predicts what happens in the clinic. PDX models, derived directly from treatment-naïve or pretreated patient tumors, retain the diversity of native tumor architecture. Studying drug response across panels of heterogeneous PDX models helps developers see which tumor types or biologically distinct variants within a disease are more likely to respond. It also reveals whether efficacy correlates with specific biomarkers, what dose ranges perform consistently across variable biology, and where resistance mechanisms are likely to emerge. Tumor diversity is not noise to be eliminated, it is a vital translational signal. Recognizing it early allows developers to refine compound selection, optimize dosing strategies, and build preclinical hypotheses that stand a better chance of holding true in patients. PDX Models as a Tool for High-Resolution Efficacy Readouts Patient-derived xenograft models offer a far more realistic view of radiopharmaceutical performance. Because they maintain heterogeneity, microenvironmental features, and in many cases prior treatment history, they give researchers a nuanced way to evaluate efficacy. For radiopharmaceuticals, PDX models deliver several advantages. They present clinically relevant antigen variability that allows researchers to assess uptake and response across a realistic spectrum of tumors. They preserve human-like tumor morphology, which helps predict intratumoral diffusion and retention. Their fidelity across passages ensures histology and molecular markers remain stable. And because study design can be adapted across tumor subtypes, expression levels, and therapeutic contexts, they enable head-to-head comparisons that are both versatile and reliable. Incorporating PDX models into efficacy studies unlocks more than traditional tumor growth inhibition curves or survival metrics. It allows teams to analyze dose-response relationships across diverse biology, track differences in duration of response, map model-specific radiosensitivity trends, and explore correlations with genomic or phenotypic biomarkers. Champions Oncology’s PDX platform builds on this by linking PDX models to clinical, genomic, and treatment response data. Tumors can even be pre-screened with tissue microarrays to identify expression-positive cases for targeted agents, streamlining model selection and aligning studies with clinical goals. For developers, the outcome is clear: studies that reflect the diversity of the patient population rather than a single optimized tumor line. Translating Efficacy Insights into Clinical Strategy Radiopharmaceuticals combine targeted delivery with localized radiation, but their success depends on early validation in models that matter. PDX-based efficacy studies don’t just generate more realistic data, they provide strategic guidance that shapes clinical development. Testing compounds across representative tumor panels reveals which patients are most likely to respond. It helps optimize dose selection and fractionation strategies by showing how retention and radiosensitivity vary across models. It uncovers resistance patterns tied to tumor phenotype, guiding combination strategies. And it strengthens IND-enabling packages by grounding them in data that reflects real-world heterogeneity. Crucially, these insights can differentiate active agents from niche responders, making trial design sharper and reducing the risk of failure from overgeneralized assumptions. Combined with biomarker data, they can even inform companion diagnostic strategies, connecting uptake and efficacy to measurable markers of patient eligibility. Conclusion: Tumor Diversity Is Not a Variable - It’s a Vital Input The path to effective, targeted radiopharmaceuticals depends on more than clever chemistry or potent payloads. It requires a clear understanding of how these agents behave across the complex biological spectrum seen in patients. PDX models, with their preserved heterogeneity and clinical relevance, offer a translational advantage that’s hard to ignore. By designing radiopharmacology studies that embrace, rather than eliminate tumor diversity, radiopharmaceutical developers can make earlier, smarter decisions that de-risk development, sharpen clinical strategy, and ultimately improve the odds of success. If you're committed to building radiopharmaceuticals that perform where it matters mos. In patients, it’s time to elevate how you evaluate efficacy. The Only CRO Pairing PDX Models with Radiopharma
Radiochemistry 101 for ADC Teams: A Practical Guide for Biodistribution Studies
In antibody–drug conjugate (ADC) development, knowing where your drug goes and whether it’s doing what you designed it to do can make the difference between success and costly setbacks. Radiochemistry offers a powerful way to generate that insight, using radioactive isotopes to “tag” antibodies, payloads, or both so their journey through the body can be tracked with precision. In this guide, we’ll break down the basics of radiochemistry for ADC teams, explain key concepts in radioisotope selection, and share practical tips to avoid common pitfalls. What Is Radioactivity and Why It Matters for ADCs Radioactivity is the process by which an unstable atomic nucleus releases energy (decay) to become more stable. For ADC developers, understanding these fundamentals is critical: Half-life (t½): The time it takes for half of the atoms to decay. This must align with your ADC’s pharmacokinetics (PK) so the isotope’s signal lasts long enough to track distribution and clearance. Decay type: Determines the detection method. For example, PET (Positron Emission Tomography) uses isotopes that emit positrons, which allows scientists to create very detailed, 3D images of how a tracer moves and accumulates in the body. SPECT (Single Photon Emission Computed Tomography) uses isotopes that emit gamma rays, producing images that show biological activity and how a drug or tracer behaves over time. Specific activity: Radioactivity per unit mass; higher values mean you can label with very little isotope, minimizing interference with binding or PK. Decay Modes and Detection Methods Different isotopes release different types of radiation, which affects how they’re used: Alpha (α): Heavy, short-range particles, mainly for therapeutic applications (e.g., Ac-225). Beta minus (β–): Electrons; common in therapeutic isotopes like Lu-177 and I-131. Beta plus (β+) / positrons: Produce PET photons for high-resolution imaging (e.g., Zr-89, Cu-64). Gamma (γ): Photons detected in SPECT imaging (e.g., In-111). Tip: Match decay type to your goal — positron emitters for imaging, beta/alpha for therapy, gamma for SPECT tracking. The Only CRO Pairing PDX Models with Radiopharma Key Concepts in Isotope Selection for ADCs Match half-life to PK: Zr-89 for long-lived antibodies; In-111 for mid-timescale studies; I-131/Lu-177 for longer courses or therapy-linked readouts. Generally, the payload should be radiolabeled with short half-life isotopes. Preserve function: Choose high-specific-activity materials and gentle conjugation chemistry. Regulatory fit & supply: Pick isotopes with robust supply chains and established handling/documentation. Imaging vs therapy: Imaging isotopes maximize detectability; therapeutic isotopes are chosen for their cytotoxic radiation. Labeling Strategies for ADCs Direct labeling (iodination) Attaches iodine isotopes (I-123/124/125/131) directly to tyrosines / histidines in the antibody. Fast and efficient but in vivo radio deiodination can occur. Residualizing tags may be used to avoid in vivo radio deiodination Indirect labeling (chelation) Uses a bifunctional chelator (e.g., DFO for Zr-89; DOTA for Lu-177) conjugated to the antibody before loading the isotope. Offers higher in vivo stability; chelator choice depends on isotope chemistry. However, although not usual, the chelators could change the PK and/or biological activity of the antibody. Payload labeling Isotope is attached to the cytotoxic payload to monitor release and clearance. Can be combined with antibody labeling (dual-label) to differentiate intact ADC from free payload. Common Pitfalls and How to Avoid Them Label instability: Choose isotope–chelator pairs with proven in vivo stability. Biological alteration: Avoid harsh labeling conditions that can impair binding or PK. PK mismatch: Don’t use a half-life that’s too short to capture late-phase distribution or clearance. Quick Reference: Isotopes for ADC Applications Isotope Half-life Emission Type Imaging Modality / Typical Use Typical Tracking Window Notes Zr-89 78.4 h (~3.3 d) β+ (positron) PET imaging – ideal for long-lived antibodies Up to 7–10 days Matches antibody PK; provides high-resolution PET images for extended studies Lu-177 6.65 d β– (beta) Therapeutic payload; can also support imaging Days to 1–2 weeks Dual-use radionuclide (therapy + imaging); strong track record in radiopharma I-131 8.02 d β–, γ (beta and gamma) Therapy and imaging for antibody/payload ADME 1–2 weeks Widely used in radioimmunotherapy; dual imaging/therapy capacity In-111 2.8 d γ (gamma) SPECT imaging – mid-timescale biodistribution 1–5 days typical (up to ~10 with cut-and-count) Best suited for 1–5 day studies; imaging resolution optimal in shorter window Ac-225 ~10 d α (alpha) Targeted alpha therapy Days to weeks (therapy-focused) Very high linear energy transfer (LET); highly cytotoxic, therapeutic only Cu-67 2.6 d β– (beta) Therapy; theranostic partner with Cu-64 Several days Can be paired with Cu-64 PET (same chemistry) for theranostic workflows Using Radiotracers in PDX and CDX Models Radiotracer studies can be performed in a range of preclinical models, but model selection directly affects how translatable your data will be. The two most common approaches for ADC biodistribution are patient-derived xenograft (PDX) models and cell line-derived xenograft (CDX) models. PDX Models What they are: Tumors from actual patients implanted into immunodeficient mice, retaining original histology and molecular characteristics. Strengths: Closely mimic human tumor biology, heterogeneity, and target expression; often more predictive of clinical outcomes. Weaknesses: More variable growth rates, higher cost, and sometimes limited availability for rare targets. CDX Models What they are: Tumors grown from established cancer cell lines implanted into mice. Strengths: Easier to grow, faster to establish, and highly reproducible; good for early proof-of-concept and method development. Weaknesses: Less heterogeneity and may not fully recapitulate the target expression or microenvironment seen in patients. Choosing the Right Model For ADC radiotracer studies, CDX models can be a cost-effective starting point to validate isotope choice and labeling chemistry, while PDX models are best for confirming biodistribution and target engagement in a clinically relevant setting before moving to the clinic. Many developers use both — starting in CDX for feasibility and scaling into PDX for translational validation. Study Design Checklist for ADC Radiolabeling Define your primary question: target engagement, linker stability, payload distribution, or therapy evaluation. Select isotope(s) to match PK and imaging/therapy needs. Choose labeling chemistry that preserves ADC function. Plan imaging modality and sampling timepoints to capture both early and late phases. Combine imaging with ex vivo biodistribution for quantitative confirmation. Include mass-dose escalation to determine receptor saturation. Radiochemistry isn’t just attaching an isotope, it’s matching the right isotope, chemistry, and study design to your ADC’s biology.

FDA Guidance: The Future of Radiopharmaceutical Development
Navigating New FDA Guidance for Radiopharmaceuticals Radiopharmaceuticals are emerging as one of the most promising frontiers in oncology, offering the ability to deliver targeted radiation directly to cancer cells while sparing healthy tissue. While the concept of using radiation in cancer therapy dates back almost a century with external beam radiotherapy, today’s systemically delivered radiopharmaceuticals represent an entirely new wave of innovation. With this promise comes new complexity, and the FDA recently released new draft guidance, Oncology Therapeutic Radiopharmaceuticals: Dosage Optimization During Clinical Development (2025), to raise the standards for how these therapies are developed and evaluated. Michael Ritchie, Chief Commercial Officer at Champions Oncology, has seen this evolution firsthand. With over a decade at Champions leading commercial operations and contributing to R&D strategy, he recognizes both the opportunity and the challenges of this emerging space. Raising the Bar: What the FDA Wants The FDA’s new guidance makes clear that more is expected of sponsors developing radiopharmaceuticals. Beyond simply demonstrating efficacy, companies must now generate robust data on pharmacodynamics, therapeutic windows, and toxicity. Regulators want to see meaningful insights into dosimetry, acute toxicities, and long-term safety profiles. They are also encouraging developers to test different doses and schedules to better define the boundaries of both safety and effectiveness. In short, the FDA is signaling that radiopharmaceuticals must be studied with the same rigor applied to other advanced oncology therapeutics, while also accounting for the unique properties of radioactive isotopes. PDX Models + Radiopharmaceuticals = Translational Power The Scientific Unknowns Despite the excitement surrounding radiopharmaceuticals, there is still much we do not know. Unlike antibody-drug conjugates, where decades of research have built a strong understanding of organ tolerability and patient management, radiopharmaceuticals remain an open field. Key questions persist: Which tumor types respond best? What are the predictable toxicities? What makes for the most effective construct from a pharmaceutical sciences perspective? Answering these questions is essential if these therapies are to fulfill their clinical potential. The Logistical Hurdles Beyond the science, logistics present another layer of challenge. Radiopharmaceuticals operate in a highly regulated space tied to nuclear medicine. Isotopes are not always easy to source, and many clinical sites lack the infrastructure or certification to administer them. This constrains patient enrollment and trial execution. Compounding the issue, isotopes have a short half-life and shelf life. Unlike traditional drugs that can be stored for months, radiopharmaceuticals must be manufactured and administered almost immediately, requiring precise planning and coordination. Charting the Path Forward The future of radiopharmaceuticals is undeniably bright, but realizing their full potential will require navigating both scientific unknowns and logistical barriers. The FDA’s Oncology Therapeutic Radiopharmaceuticals guidance provides structure, but it also highlights the complexity of the journey ahead. Success will depend on deeper biological insights, smarter trial design, and operational excellence in manufacturing and delivery. The Only CRO Pairing PDX Models with Radiopharma At Champions Oncology, we are committed to partnering with pharma and biotech innovators to address these challenges head-on. By leveraging our advanced preclinical models and translational platforms, we help generate the data and confidence needed to accelerate radiopharmaceutical development and bring these groundbreaking therapies closer to patients.

Radiopharmaceuticals in Cancer Treatment: Insights from Mike Ritchie
In a recent video interview with Pharmaceutical Technology, Mike Ritchie, Chief Commercial Officer at Champions Oncology, shared his perspective on the fast-evolving field of radiopharmaceuticals. Drawing on his 20 years of experience in cancer research and leadership roles at Pfizer and Champions, Mike highlighted what makes radiopharmaceuticals unique, the challenges they present, and how advanced models and real-world data are shaping their development. Q: What is unique about the use of radiopharmaceuticals to treat cancer? Radiopharmaceuticals have actually been around for nearly a century. In the early days, radiation was directed externally using large machines to treat tumors. Over time, the technology evolved to address internal tumors as well. What makes today different is the transformation inspired by antibody-drug conjugates (ADCs). ADCs showed us that toxic payloads could be delivered precisely to tumors, sparing healthy tissue. After decades of learning how to design and manufacture ADCs effectively, we now understand what makes a targeted therapy work. Radiopharmaceuticals are taking a similar approach. By linking radioactive isotopes—what Mike calls “radio ligands”—to antibodies, scientists can deliver radiation directly to cancer cells. This creates radio-drug conjugates, an exciting new mechanism of action that could address unmet needs, expand the therapeutic index, and potentially offer safer treatment options for patients. PDX Models + Radiopharmaceuticals = Translational Power Q: What are the challenges associated with developing radiopharmaceuticals? The hurdles fall into two categories: drug development and tumor biology. From a development standpoint, researchers are still learning how to design radiopharmaceuticals that are stable, manufacturable at scale, and behave predictably in patients. Linking a radioligand to an antibody is a complex chemistry challenge, and improving these linkers remains a key opportunity for the field. Biologically, we are just beginning to understand which tumor types and molecular subgroups are most responsive. As with other therapies, some cancers will respond while others develop resistance. Identifying the right patient cohorts will be critical as these drugs advance through clinical development. Q: How are PDX models used in radiopharmaceutical testing? Patient-derived xenograft (PDX) models play a central role in this area. They allow researchers to test radiopharmaceuticals in tumor models that more closely mirror the clinical setting. Patients in clinical trials are often heavily pretreated, with metastatic disease across multiple organs. These advanced lesions are the ones that ultimately determine patient outcomes, so therapies must be tested against representative biology. At Champions, large PDX libraries are used to simulate clinical trials in animals, testing drugs across diverse tumors. The results not only reveal where a therapy works, but also highlight the molecular features linked to response or resistance. This helps guide patient selection strategies for clinical trials. Q: What role does real-world data play in radiopharmaceutical development? Real-world data provides critical context: how patients are treated, how they respond, the stage and metastatic nature of their disease. But Mike emphasizes that pairing this clinical information with deep molecular data is where the real value lies. Champions is unique in combining these two layers. Their datasets link real-world patient outcomes with rich tumor profiling, including whole-exome sequencing, RNA-seq, proteomics, phospho-proteomics, and cell surface proteomics. This combination allows researchers to uncover vulnerabilities in tumors, predict resistance mechanisms, and design smarter clinical trials. When companies run simulated trials at Champions, they can leverage this integrated dataset to refine predictions and improve translational relevance. Closing Thoughts As radiopharmaceuticals move from concept to clinic, success will depend on solving both development and biology challenges. The lessons learned from ADCs are accelerating progress, but identifying the right patients and optimizing drug design remain key hurdles. By combining advanced preclinical models with integrated real-world and molecular data, Champions Oncology is helping researchers de-risk development and bring this promising new class of therapies closer to patients who need them most. The Only CRO Pairing PDX Models with Radiopharma
Seeing Beyond Efficacy: How Radiolabeling Advances ADC Drug Development
Antibody-drug conjugates (ADCs) have transformed oncology by combining targeted delivery with potent cytotoxic payloads. But traditional preclinical studies, focused on tumor growth inhibition, often leave blind spots in understanding how these complex molecules behave in the body. That’s where radiolabeling changes the game. In our recent webinar, Radiotracers and ADC Targeting: Scientific Insights for In Vivo Validation and Clinical Transition, experts Dr. Denis R. Beckford-Vera (Champions Oncology) and Dr. Shankar Vallabhajosula (Weill Cornell Medicine, Convergent Therapeutics) unpacked the science and strategy behind using radiotracers to generate decision-driving data for ADC development. WEBINAR Radiotracers and ADC Targeting: Scientific Insights for In Vivo Validation and Clinical Transition Why Radiolabeling Matters for ADCs Unlike unconjugated antibodies, ADCs can vary significantly in pharmacokinetics, biodistribution, and biotransformation depending on conjugation methods, linker chemistry, and payload properties. These factors directly influence efficacy and safety. Radiolabeling both the antibody and/or payload enables you to: Quantify tissue distribution across the whole body Assess target engagement in tumors versus off-target organs Predict efficacy earlier in development Measure payload release and clearance for linker optimization Key Takeaways from the Webinar 1. Dual Radiolabeling Reveals the Full Picture By attaching one isotope to the antibody (e.g., Zr-89) and another to the payload (e.g., I-131), researchers can simultaneously track both components without signal interference. This approach reveals where the conjugate stays intact and where the payload is released, a critical insight for linker stability and safety profiling. 2. Imaging as a Predictor of Response Molecular imaging with Zr-89–labeled ADCs has shown that tumor uptake patterns can predict which preclinical models respond to therapy. In responsive models, tumor-to-blood ratios are higher and sustained, while non-responsive models show uptake similar to non-specific controls. 3. Dual-Mechanism Therapeutic Potential Pairing a radiolabeled antibody with a chemotherapeutic payload in a single ADC can produce additive or even synergistic anti-tumor effects, as demonstrated with Lu-177 + MMAE combinations. 4. Translating Preclinical Insight to the Clinic In a HER3-targeting immuno-PET study, Zr-89–labeled antibodies were used to measure dose-dependent target engagement in patients. Imaging revealed saturable tumor uptake at higher mass doses, confirming target specificity and informing dose optimization strategies—insights not possible from biopsy alone. The Champions Oncology Advantage Champions Oncology integrates radiochemistry, in vivo imaging, biodistribution, and PDX model expertise into a streamlined workflow. This enables ADC developers to: Validate tumor targeting in clinically relevant models Quantify on-target and off-target uptake with precision Generate high-quality biodistribution and PK data to support IND submissions and partner discussions Ensure secure isotope supply for uninterrupted studies Watch the Full Webinar The blog you’re reading is just the highlights. In the full webinar, Radiotracers and ADC Targeting: Scientific Insights for In Vivo Validation and Clinical Transition, Dr. Beckford-Vera and Dr. Vallabhajosula dive into: Radiochemistry techniques for antibodies and payloads Case studies from approved and experimental ADCs Isotope selection and chelation strategies Translational use cases bridging preclinical and clinical studies Watch the full recording to see how radiolabeling can de-risk your ADC program, accelerate timelines, and improve the odds of clinical success. Watch the Webinar: Radiotracers and ADC Targeting
How Radioisotopes Are Being Used to De-Risk Antibody-Drug Conjugate (ADC) Development
Antibody-drug conjugates (ADCs) continue to generate momentum in oncology, offering the promise of targeted cytotoxic delivery with greater specificity and reduced systemic toxicity. However, the complexity of these molecules, from antigen binding and internalization to payload release and clearance, makes them notoriously difficult to optimize. Many ADC programs stall not because the payload is ineffective, but because developers lack insight into whether the antibody is reaching and engaging the target in vivo. Radiotracers offer a unique opportunity to bridge this knowledge gap. By radiolabeling antibodies, , or ADC-like constructs with radioisotopes, researchers can evaluate in vivo biodistribution, tumor targeting, and off-target accumulation long before clinical trials. These tools provide a non-invasive, high-resolution view of compound behavior that traditional pharmacokinetic assays or histology cannot capture alone. In this article, we explore how radiotracers techniques, when combined with clinically relevant preclinical models, are helping ADC developers de-risk critical decisions earlier in the development cycle. From model selection to target validation to biodistribution profiling, radiotracers are becoming an indispensable part of the ADC toolkit. PDX Models + Radiopharmaceuticals = Translational Power Understanding Risk in ADC Development The appeal of ADCs lies in their elegant concept: deliver a cytotoxic payload directly to cancer cells via a highly specific antibody, sparing healthy tissue. In practice, however, ADC development is fraught with failure points. Many candidates show limited efficacy or unacceptable toxicity — not due to poor payload design, but because of incomplete understanding of how the construct behaves in vivo. Key risks include: Heterogeneous or insufficient target expression, which leads to poor tumor uptake Off-target accumulation in antigen-expressing normal tissues Suboptimal pharmacokinetics, including premature clearance or payload release Lack of internalization or poor intracellular trafficking, reducing payload delivery These risks are difficult to detect with traditional in vitro methods alone. Even when early pharmacology data appear favorable, translational failures often emerge when the compound enters more physiologically complex systems or encounters unexpected biological variability. What ADC developers need is a translational lens into antibody behavior — one that captures how the full construct distributes across tumor and normal tissue, how long it persists at the target site, and how that behavior varies across models. That’s where radiotracers come in. Radiolabeling ADCs to Visualize Biodistribution and Target Engagement Radiolabeling ADCs with radioisotopes enables developers to evaluate compound behavior with far greater granularity than traditional methods. By tagging the antibody or the full ADC construct with an isotope such as Zirconium-89 (Zr-89), Lutetium-177 (Lu-177) or Indium-111 (In-111), researchers can track distribution, uptake, and retention across tissues and tumor models in real time. This technique serves multiple functions: Biodistribution profiling: Understand where the antibody accumulates, and how much it localizes to the tumor versus healthy organs Target engagement assessment: Confirm that the biologic is reaching antigen-positive tumors with sufficient intensity and duration In vivo tumor targeting characterization in well characterized PDX models: Understand ADC distribution variability across PDX with preserved inter- and intra-tumoral heterogeneity found in human cancers. Off-target surveillance: Identify unintended uptake in antigen-expressing normal tissues (e.g., liver, spleen, bone marrow) early in development (depending on cross reactivity) Comparative evaluation: Screen multiple antibodies, formats, or linkers to determine which offers the best tumor-to-background ratio Radiolabeled ADC constructs can be studied across multiple timepoints to assess dynamic distribution and clearance profiles. When paired with well-characterized preclinical models—particularly those that reflect human heterogeneity in target expression, this approach allows for an evidence-based refinement of lead selection and study design. In addition to guiding compound optimization, radiolabeled compounds generate data that supports IND-enabling work by illustrating tumor specificity and helping predict potential toxicity risks related to off-target delivery. Using PDX Models to Reflect Real-World Variability in Antigen Expression While radiolabeling provides a powerful tool for visualizing distribution, the choice of model system ultimately determines how meaningful those insights will be. Cell line–derived xenografts (CDX), though commonly used, often overexpress the target antigen in a uniform and artificial manner. This can mask important limitations in targeting specificity and distribution, leading to false confidence in a compound’s performance. In contrast, patient-derived xenograft (PDX) models preserve the inter- and intra-tumoral heterogeneity found in human cancers. Differences in antigen density, vascularization, stromal composition, and tumor architecture all impact how an ADC or radiolabeled construct will behave in vivo. Testing compounds across a panel of PDX models allows developers to assess performance across a range of real-world tumor phenotypes, gaining visibility into variability that may influence clinical response. Champions Oncology’s Lumin platform includes hundreds of PDX models annotated with genomic, phenotypic, and treatment-response data. These models can be screened in advance using tissue microarrays (TMAs) to identify tumors with varying levels of antigen expression, enabling strategic model selection and rational study design. In the context of radiopharmaceuticals or radiolabeled ADCs, this means developers can: Evaluate targeting across low, mid, and high-expressing tumors Identify models most likely to mirror patient response Explore relationships between uptake and known molecular drivers By integrating radiolabeling with clinically annotated, heterogenous models, developers gain a more complete picture of how a compound is likely to perform across the clinical population —and avoid late-stage surprises. From Imaging to Strategy: De-Risking ADC Development Earlier in the Pipeline The high cost and complexity of ADC development demand early, informed decision-making. Traditional pharmacology and histology provide critical insights, but they don’t tell the whole story, especially when it comes to understanding how a biologic behaves in real biological systems over time. Radiotracers tools fill that gap by enabling non-invasive, temporal, and quantitative evaluation of ADC behavior across diverse, clinically relevant models. When used early in development, radio labeling and biodistribution studies can help developers: Select better antibody constructs or formats based on real in vivo performance Prioritize linker-payload combinations with favorable pharmacokinetics and tumor retention Predict potential toxicity or dosing issues from off-target accumulation Justify model selection and dose rationale in regulatory submissions By integrating these insights into the design phase — not as a retrospective check — developers can refine their therapeutic strategy while reducing attrition risk later in the pipeline. Radiotracers not only illuminate compound behavior but also serve as a translational bridge that connects target biology, delivery, and clinical feasibility. In a field where timelines are long, investment is high, and failure is costly, this approach offers a pragmatic and data-rich path forward: study smarter, screen earlier, and develop with confidence. The Only CRO Pairing PDX Models with Radiopharma
.jpg)
Why PDX Models Are Essential for Radiopharmaceutical Testing
Radiopharmaceuticals represent a rapidly advancing class of targeted oncology therapeutics, leveraging radionuclide-labeled molecules to deliver ionizing radiation directly to tumor cells. Despite the promising clinical potential of alpha- and beta-emitting radiopharmaceuticals, achieving translational success remains challenging. Robust, well-characterized preclinical models are essential to increase confidence in compound performance before entering the clinic. Where biodistribution, receptor heterogeneity, and tumor penetration critically influence therapeutic index and patient selection strategies, traditional preclinical models often fall short. Cell line–derived xenografts (CDX), in particular, offer limited predictive value due to their clonal homogeneity, uniform tumor architecture, and lack of biological diversity, factors that can lead to inaccurate assessments of targeting performance, distribution, and treatment efficacy. Patient-derived xenograft (PDX) models offer a superior alternative, retaining the histological architecture, molecular diversity, and intra, and inter-tumoral heterogeneity of the donor patient’s tumor. These attributes enable more physiologically relevant assessment of targeting efficacy, radiotracer distribution, and therapeutic response—key metrics in determining compound viability prior to clinical translation. In this article, we examine the limitations of traditional models, the biological advantages of PDX platforms, and the specific ways in which PDX enhances radiopharmaceutical study design. We also highlight how access to large, clinically annotated model libraries—such as Champions Oncology’s Lumin platform can support more informed, data-driven decisions during preclinical development. PDX Models + Radiopharmaceuticals = Translational Power The Limits of Traditional Preclinical Models Despite their ubiquity in oncology research, traditional preclinical models—particularly cell line–derived xenografts (CDX)—present significant limitations for targeted drug development, including radiopharmaceuticals. CDX models are generated by implanting immortalized cancer cell lines into immunodeficient mice. While they offer logistical advantages such as rapid tumor growth and reproducibility, these models are inherently reductionist. Their clonal architecture lacks the genomic and phenotypic heterogeneity observed in primary tumors, which can lead to misleading conclusions regarding target accessibility, tumor penetration, and intratumoral uptake of radiolabeled compounds. Moreover, CDX models typically fail to recapitulate the complex tumor microenvironment (TME), including stromal interactions, vasculature, and immune contexture—all of which are known to influence radiopharmaceutical distribution and efficacy. In addition, receptor expression in cell lines is often artificially uniform or overexpressed, providing an inaccurate representation of clinical target variability. For radiopharmaceuticals — where therapeutic performance depends heavily on fine, tuned targeting, localized retention, and clearance kinetics, these simplifications are not benign. Data generated from CDX models may overestimate therapeutic potential or fail to predict safety liabilities, contributing to a translational gap between preclinical validation and clinical outcomes. What Makes PDX Models Different Patient-derived xenograft (PDX) models are established by implanting primary tumor tissue directly from oncology patients into immunodeficient mice, preserving the cellular heterogeneity, stromal components, and histopathological architecture of the original tumor. Unlike CDX systems, PDX models retain critical aspects of human tumor biology across multiple passages. This biological fidelity translates into substantial advantages for radiopharmaceutical development. First, PDX models capture both intratumoral and intertumoral heterogeneity, a key determinant of response variability in radiolabeled therapies. Differences in antigen density, receptor expression, vascularization, and stromal composition can significantly affect radiotracer uptake and therapeutic distribution—elements that are often uniform or absent in traditional systems. Second, because PDX tumors grow in vivo without prior dissociation or in vitro manipulation, their tumor microenvironments more accurately reflect the spatial and structural complexity of human malignancies. This includes irregular vasculature, hypoxic regions, and heterogeneous interstitial pressure—factors that influence compound diffusion, radiation deposition, and biological effects. PDX models have demonstrated greater predictive validity than CDX systems across multiple drug classes, with treatment responses that more closely reflect clinical outcomes. This makes them particularly valuable for de-risking radiopharmaceutical assets in the early stages of development, providing insight into variability in target engagement and therapeutic effect. While tumor-specific uptake can be assessed in a human-relevant context, off-target distribution in preclinical models may not fully reflect human cross-reactivity due to interspecies differences in antigen expression. In short, PDX models offer a translational bridge between mechanistic discovery and clinical decision making, one that is especially important when developing complex, spatially dependent therapies like radiopharmaceuticals. How PDX Enhances Radiopharmaceutical Testing The evaluation of radiopharmaceuticals requires more than evidence of cytotoxicity; it demands a nuanced understanding of how a radiolabeled compound distributes within and interacts with—a tumor and its microenvironment. PDX models provide the translational resolution needed to interrogate these complex dynamics. One of the primary advantages of using PDX models in this context is the ability to model inter-patient variability in target expression. Radiopharmaceuticals often rely on the presence of specific cell surface antigens or receptors for tumor localization. In a clinical setting, these markers are rarely expressed uniformly across patient populations. By leveraging a library of diverse PDX models—each with distinct molecular and phenotypic profiles—researchers can assess how differences in target expression influence uptake, specificity, and efficacy. Additionally, PDX models enable realistic biodistribution analysis in tumors that replicate human heterogeneity in vascular density, stromal content, and perfusion. These factors play a significant role in modulating the intratumoral deposition of radiolabeled compounds, Preclinical studies in PDX therefore allow developers to anticipate challenges related to tracer penetration, off, target accumulation, and clearance kinetics, challenges that CDX models routinely obscure. Efficacy evaluation is another area where PDX models offer substantial value. Because these tumors respond to treatment in ways that reflect clinical patterns, including partial response, acquired resistance, and heterogeneous regression, they offer a more realistic basis for determining therapeutic window, optimal dosing, and potential biomarkers of response. When used systematically, PDX models allow radiopharmaceutical developers to move beyond binary efficacy readouts and instead generate layered, clinically relevant insights into compound behavior—insights that inform both development decisions and regulatory discussions. The Lumin Advantage While the value of PDX models in radiopharmaceutical development is clear, the ability to scale these insights depends on access to a diverse, well-characterized model library. Champions Oncology’s PDX platform is the most deeply annotated and clinically relevant PDX collections available globally, enabling sponsors to tailor studies with unprecedented precision. The library encompasses thousands of PDX models derived from a wide range of solid tumors, each backed by comprehensive clinical, histological, and molecular data. This includes mutational profiles, gene expression signatures, prior treatment history, and, critically for radiopharmaceutical programs, data on target expression heterogeneity across tumor types. To accelerate the design of rational studies, complementary tissue microarrays (TMAs) prepared from the PDX collection are also available. These arrays allow researchers to screen panels of models for antigen expression or biomarker prevalence prior to initiating in vivo work, enabling efficient model selection, improving study design, and reducing downstream variability. In radiopharmaceutical testing, where variability in receptor density or antigen availability can dramatically influence tracer uptake and therapeutic effect, this level of pre-screening and data integration is a strategic advantage. It allows developers to assess compound performance across diverse biological backgrounds and identify model subsets most likely to inform clinical translation. Combined with Champions’ in-house imaging, conjugation, and radiolabeling capabilities, Lumin platform offers a comprehensive ecosystem for generating radiopharmaceutical data that’s not only robust—but truly relevant to human disease. The Only CRO Pairing PDX Models with Radiopharma

Beyond TCGA: TumorGraft’s New Frontier in Cancer Research
Imagine a world where doctors can predict how a tumor will respond to treatment before a patient starts therapy. For decades, cancer researchers have relied on The Cancer Genome Atlas (TCGA), a massive dataset of 20,000 samples across 33 cancer types, to decode the molecular secrets of tumors. But TCGA has a blind spot: it mostly studies untreated, primary tumors, leaving critical questions about advanced cancers and treatment responses unanswered. Enter the Champions Oncology TumorGraft® platform—a game-changer in cancer research. With 1,500 patient-derived tumor models from over 50 cancer types, TumorGraft® captures the real-world complexity of advanced, metastatic, and heavily treated tumors. By combining molecular data with detailed pretreatment histories and treatment response insights, it’s opening new doors for predicting drug effectiveness, uncovering resistance mechanisms, and designing smarter therapies. In this post, we’ll explore how TumorGraft® complements TCGA, using a fascinating case study on mutational signatures to show its power. TCGA: The Gold Standard with Limits TCGA is a cornerstone of cancer research. It's publicly accessible, high-quality data, spanning DNA, RNA, and proteins, has fueled countless discoveries about how tumors develop. Researchers use it to characterize tumors, improve diagnoses, and identify molecular drivers of cancer. But TCGA isn’t perfect. Most of its samples come from primary, untreated tumors, with only a small fraction (about 42 patients) having received neoadjuvant treatment. This makes TCGA ideal for studying cancer’s early stages but less useful for advanced, metastatic, or post-treatment tumors. It also underrepresents certain populations and cancer stages, lacks pretreatment histories, and doesn’t allow access to physical samples for follow-up experiments. If you’re studying treatment resistance or real-world patient outcomes, TCGA’s data can only take you so far. TumorGraft®: A Window into Advanced Cancers The TumorGraft® platform, developed by Champions Oncology, flips the script. Its 1,500 patient-derived xenografts (PDXs)—tumors grown in mice to mimic human cancer—represent over 50 cancer types, focusing on advanced-stage, metastatic, and pre-treated tumors. These models reflect the diversity and complexity of patients seen in clinics, where cancers often evolve under the pressure of multiple therapies. Unlike TCGA, TumorGraft® includes detailed pretreatment information, capturing the therapies patients received before their tumors were sampled. This data, which can be further mined, offers a window into how prior treatments shape tumor biology, enabling researchers to study real-world clinical scenarios. Combined with treatment response data from in vivo experiments, where Champions Oncology has tested drugs representing multiple standards of care and measured Tumor Growth Inhibition (TGI)—how much a drug slows tumor growth—TumorGraft® unlocks use cases TCGA can’t touch: • Predicting Treatment Responses: See how a tumor’s molecular profile and pretreatment history predict its reaction to specific drugs. • Discovering Biomarkers: Identify markers that signal whether a treatment will work, informed by prior therapies. • Understanding Resistance: Study why some tumors resist therapy and find ways to overcome it, leveraging pretreatment data. • Improving Combination Therapies: Test drug combinations to find the most effective mixes, considering treatment histories. • Finding New Drug Targets: Link molecular features and pretreatment patterns to treatment outcomes to uncover novel therapies. Mutational Signatures: A Shared Language To prove TumorGraft’s® reliability, researchers compared it to TCGA using mutational signatures—DNA damage patterns that act like fingerprints, revealing what caused a tumor, like UV light or faulty DNA repair. These signatures, cataloged in the COSMIC database, help identify vulnerabilities in tumors and guide treatment strategies. The analysis began by curating COSMIC signatures and calculating their exposure and frequency in both TCGA and TumorGraft® datasets. Figure 1: Diagram showing calculated exposure/frequency of tumors in TumorGraft® (left) and TCGA (right). The results were striking. Both datasets flagged similar patterns. For example, SBS7a—a signature tied to UV light exposure (COSMIC SBS7a)—was strongly linked to melanoma in both TumorGraft’s® PDX models and TCGA’s samples. Analysis of 1,155 PDXs revealed SBS7a as a hallmark of melanoma, mirroring TCGA’s findings. This alignment shows that TumorGraft’s® data is as biologically accurate as TCGA’s, despite its focus on advanced, treated tumors. Figure 2: Chart of multi-omic data analysis from 1,155 TumorGraft® PDXs, highlighting trends in mutational signatures across tumor types. Figure 3: Bar chart showing SBS7a mutational signature prevalence in melanoma PDXs from TumorGraft®, with high association compared to other cancer types. Researchers analyzed signatures using different motifs—single base substitutions (SBS96), double base substitutions (DBS78), and insertions/deletions (IND83). All signatures found in TumorGraft® appeared in TCGA, with TCGA’s larger sample size revealing a few extra patterns due to its scale. Figure 4: Comparison of mutational signatures across SBS96, DBS78, and IND83 motifs in TumorGraft® and TCGA datasets. A UMAP plot, a visual tool that maps data similarity, confirmed that tumors cluster by type (e.g., melanoma, lung), not by whether they came from TCGA or TumorGraft®. This suggests that biology, not the data source, drives the differences—a green light for using TumorGraft® alongside TCGA. Figure 5: UMAP scatterplot showing tumors clustered by type, not data source, confirming TumorGraft’s® biological consistency with TCGA. Figure 6: Dot plot showing associations between mutational signatures and metadata, with dot size indicating event frequency and color representing p-value. Notably, tumors with signatures SBS6, SBS15, and SBS10b showed resistance to alkylating agents and platinum-based chemotherapies, exhibiting lower TGI. According to COSMIC, SBS6 and SBS15 are linked to defective DNA mismatch repair, common in microsatellite-unstable tumors, while SBS10b is tied to mutations in DNA polymerase epsilon, often seen in hypermutator tumors. By mining pretreatment data, researchers can explore how prior therapies influence these resistance patterns, offering clues to personalize treatments and avoid ineffective drugs. Figure 7: Graph showing correlation between SBS6, SBS15, and SBS10b signatures and resistance to alkylating agents/platinum in TumorGraft® PDXs, with lower tumor growth inhibition (TGI). This is just the beginning. TumorGraft’s® pretreatment data, combined with patient treatment histories and in vivo responses to thousands of standard-of-care drugs, is a goldmine for studying how prior therapies shape tumor evolution and treatment outcomes. Why TumorGraft® Matters The similarities between TCGA and TumorGraft’s® mutational signatures prove that TumorGraft® is a reliable partner to the gold standard. But its focus on advanced, treated tumors, detailed pretreatment information, and treatment response data takes cancer research to new heights. The ability to mine pretreatment histories—unavailable in TCGA—enables researchers to uncover how past therapies influence tumor biology, paving the way for personalized medicine and better patient outcomes. Whether you’re developing new drugs, tackling treatment resistance, or designing combination therapies, TumorGraft® provides insights that TCGA can’t. Looking ahead, researchers can dig deeper with TumorGraft® by exploring copy number signatures, RNA sequencing, or proteomics. Mining pretreatment data alongside these analyses could reveal even more about how tumors evolve under therapeutic pressure, driving breakthroughs in cancer care. Ready to Transform Cancer Research? The TumorGraft® platform is more than a dataset—it’s a bridge to personalized medicine. By combining the molecular depth of TCGA with TumorGraft’s® real-world treatment and pretreatment insights, researchers can unlock answers that bring us closer to curing cancer. Want to explore TumorGraft® for your next study? Learn how our platform can power your research today. Note: Data sourced from Champions Oncology and validated against TCGA’s data using the mutational motifs in COSMIC.
Predicting ADC Efficacy Using IHC and NGS
Antibody-drug conjugates (ADCs) represent a cutting-edge advancement in cancer therapy. These unique biopharmaceuticals act as "smart bombs," combining monoclonal antibodies specifically targeting cancer cells with potent cytotoxic drugs delivered directly to the tumor site. This precise targeting reduces collateral damage to healthy cells, minimizing adverse effects. Given the growing adoption of ADCs in clinical oncology, predicting their efficacy has become a critical challenge. Factors such as tumor heterogeneity, antigen expression, and individual patient differences underscore the need for precise biomarkers and advanced tools to determine patient suitability. The integration of immunohistochemistry (IHC) and next-generation sequencing (NGS) has emerged as a powerful approach to refining this prediction process. This blog explores the challenges of predicting ADC efficacy, the roles of IHC and NGS, and how these technologies are shaping the future of ADC-based therapy. The Importance of Predicting ADC Efficacy Why Predicting ADC Outcomes is Crucial? The therapeutic landscape of ADCs continues to evolve, with several FDA-approved ADCs and many others progressing through clinical trials. However, not all patients with cancer respond to these therapies, making the prediction of ADC efficacy vital to ensuring optimal outcomes. Key considerations include: • Target Antigen Expression: ADCs' performance depends on the presence and density of specific antigens on tumor cells. • Tumor Heterogeneity: Variability in antigen expression within and between tumors can impact ADC penetration and effectiveness. • Resistance Mechanisms: Both primary and acquired resistance to ADCs challenge their sustained efficacy. The Role of Immunohistochemistry in Predicting ADC Efficacy How IHC Works in ADC Therapy? Immunohistochemistry (IHC) is a gold standard for detecting protein expression within tumors. By applying antigen-specific antibodies to tissue samples, IHC enables visualization and quantification of target antigens. For ADCs, this method is highly valuable in determining whether a patient’s tumor expresses the antigen necessary for ADC binding and delivery. Benefits of Using IHC for ADC Target Assessment • Direct Visualization: Precise localization of target antigens not only confirms presence but also identifies antigen distribution within the tumor. • Threshold Analysis: IHC enables clinicians to set expression thresholds for ADC targeting, ensuring that only eligible patients receive therapy. • Readily Available Tool: IHC is widely accessible across pathology labs, making it a practical option for many cancer centers. Challenges in IHC Analysis Quantification of protein expression in IHC is typically assessed with H-scores, calculated by pathologists based on the identification of the percentage of cancer cells expressing the target and its level of intensity. The subjectivity of the methodology is an inherent risk of inconsistency for inter- intra- assay, for this reason, the H-score is usually calculated as the result of the independent IHC data analysis from at least two pathologists. Also, despite its benefits, IHC has limitations in predicting ADC efficacy, as for known ADC targets such as HER2 and TROP2, the correlation between target expression-related IHC scores and ADC efficacy, is not always strong. Next-Generation Sequencing (NGS) Contributions to ADC Accuracy What is NGS and How it enhances ADC Targeting? Next-generation sequencing (NGS) analyzes DNA, RNA, and gene expression at unprecedented speed and precision. By providing data-rich insights, NGS enables oncology researchers to evaluate molecular profiles in addition to traditional methods like IHC. In particular, NGS data can help researchers with the identification of biomarkers that may predict ADC responses. Recent studies have demonstrated NGS's advantages in ADC biomarker identification. For example, RNA sequencing correlations with IHC staining (e.g., TROP2, HER2) highlight strong alignment in certain targets, offering the potential for RNA-based cutoffs to complement IHC in ADC prediction. Variability, however, remains for some antigens, underscoring the need for continuing refinement. Innovations in Predicting ADC Efficacy Beyond IHC and NGS Emerging technologies and methodologies are refining ADC efficacy prediction even further. Key innovations include: • Multivariate Biomarkers: Next-gen tools like ADC Treatment Response Scores (ADC-TRS) evaluate gene expression alongside additional factors (e.g., adhesion, proliferation markers), significantly enhancing response prediction. • AI-Powered Pathology: Artificial intelligence in cancer pathology is enabling automated image and molecular data analysis, providing deeper insights into tumor heterogeneity and antigen expression thresholds. • Molecular Imaging: Imaging technologies are being integrated with NGS and IHC to provide real-time visualization of ADC biodistribution within patient tumors. Future Potential • Predictive Precision: Enhanced tools and algorithms will improve patient stratification, leading to better survival outcomes and fewer treatment-related toxicities. • Adaptive Therapies: With the ability to monitor antigen dynamics over time, clinicians can tailor ADC therapies to evolving tumor characteristics. Accurate Predictions Mean Better Outcomes for Patients Antibody-drug conjugates are paving the way for highly targeted and effective cancer treatments. However, maximizing their potential hinges on the ability to accurately predict suitable candidates through methods like IHC and NGS. By leveraging the latest advancements in predictive biomarkers and sequencing technologies, scientists and oncologists can improve patient outcomes, advancing precision medicine to new heights. As the field evolves, innovations will continue to refine ADC efficacy predictions, enabling personalized treatment strategies that benefit patients across diverse cancer types. Reach out to Champions Oncology to learn more about how we can help you develop your ADCs with our cutting-edge ex vivo and in vivo platforms and predictive tools that drive innovation. [1] Katrini J, Boldrini L, Santoro C, Valenza C, Trapani D, Curigliano G. Biomarkers for Antibody-Drug Conjugates in Solid Tumors. Mol Cancer Ther. 2024 Apr 2;23(4):436-446. doi: 10.1158/1535-7163.MCT-23-0482. PMID: 38363729. [2] Sachdev P. Thomas, Laurel A. Habel, Jennifer Marie Suga, Ninah Achacoso, Josh Nugent, Katarina M. Robinson, Ryan White, and Scott A. Tomlins. Evaluation of a predictive biomarker for antibody drug conjugates (ADCs). Journal of Clinical Oncology, Volume 42, Number 16_suppl. doi.org/10.1200/JCO.2024.42.16_suppl.3140 [3] Makawita S, Meric-Bernstam F. Antibody-Drug Conjugates: Patient and Treatment Selection. Am Soc Clin Oncol Educ Book. 2020 Mar;40:1-10. doi: 10.1200/EDBK_280775. PMID: 32213087. [4] Kushnarev V, Stupichev D, Kryukov K, et al143 Correlating RNA-seq detection and IHC staining of potential antibody-drug conjugate (ADC) targets: HER3, HER2, TROP2, Nectin4, and aFLRJournal for ImmunoTherapy of Cancer 2023;11:doi: 10.1136/jitc-2023-SITC2023.0143 [5] Ascione L, Crimini E, Trapani D, Marra A, Criscitiello C, Curigliano G. Predicting Response to Antibody Drug Conjugates: A Focus on Antigens' Targetability. Oncologist. 2023 Nov 2;28(11):944-960. doi: 10.1093/oncolo/oyad246. PMID: 37665782; PMCID: PMC10628585. [6] Paolo F. Caimi, Mehdi Hamadani, Carmelo Carlo-Stella, Masoud Nickaeen, Eric Jordie, Kiersten Utsey, Tim Knab, Francesca Zammarchi, Serafino Pantano, Karin Havenith, Ying Wang, Joseph Boni; CD19 Expression by IHC Alone Is Not a Predictor of Response to Loncastuximab Tesirine: Results from the LOTIS-2 Clinical Trial and Quantitative Systems Pharmacology Modeling. Blood 2022; 140 (Supplement 1): 9548–9550. doi: https://doi.org/10.1182/blood-2022-159626
Advancements in bispecific antibody research and innovative platforms
Bispecific antibodies represent a groundbreaking innovation in cancer immunotherapy. By binding to two distinct antigens simultaneously, these molecules direct immune cells to target cancer cells with precision, enhancing immune-mediated killing. The development of these therapies has opened new frontiers in oncology, with preclinical platforms playing a pivotal role in evaluating their efficacy, safety, and mechanisms of action before clinical trials. History and Challenges of Bispecific Antibodies Research The concept of bispecific antibodies originated in the 1960s, with the first experimental bispecific constructs developed in the 1980s using chemical cross-linking. Early versions were limited by instability and production challenges. Advances in genetic engineering in the 1990s led to the creation of more stable and functional bispecific molecules. Over time, the field has evolved to include trispecific, tetraspecific, and even pentaspecific antibodies, each designed to address specific therapeutic needs by engaging multiple targets or pathways simultaneously. The structural complexity of these molecules has grown, incorporating modular designs like dual-variable domain antibodies and Fc-engineered formats to enhance therapeutic efficacy and half-life. [1, 2] The dual mechanisms of bispecific antibodies pose unique challenges in preclinical research. Traditional cell line-based models often fail to replicate the intricate interactions within the tumor microenvironment (TME), limiting their predictive power. Advanced models that closely mimic human tumor biology are required to fully understand the therapeutic potential and risks of bispecific antibodies. Testing Bispecific Antibodies: In Vivo and Ex Vivo Approaches Advanced preclinical models have been used to evaluate the performance of bispecific antibodies. Among the pre-clinical models, patient-derived xenografts (PDX) are invaluable for maintaining the genetic and histological fidelity of patient tumors. Representing the testing platform closer to the clinic, these models allow researchers to study tumor-specific responses and resistance mechanisms in vivo. Syngeneic models, using murine tumors in immunocompetent mice, provide insights into the immune system’s role in therapy efficacy. Yet, murine tumors and immune responses are very different from their human counterpart. While syngeneic models are a good proof of concept, they do not provide clinically translatable reliable data sets. [3] Humanized mice incorporating human tumors together with human immune cells are very important in the field. The standard models used to test bispecific are adoptive transfer models leveraging peripheral blood mononuclear cells (PBMCs) or CD34+ stem cells. These systems replicate the interactions within the TME, enabling the study of immune cell recruitment, activation, and cytotoxicity driven by bispecific antibodies. The integration of humanized mouse models, engrafted with PBMCs or CD34+ stem cells, enhances the translational relevance of these studies by mimicking human immune responses more accurately. The use of humanized mice models can be challenging; however, the right expertise can help the selection of the best strategy for each bispecific MoA or combinatorial treatment of interest with CPI. [4] Organoid models offer a controlled ex vivo environment to study complex cellular interactions, preserving the molecular diversity and histological characteristics of original patient tumors. Adding immune cells to organoids generating a co-culture assay enables researchers to analyze interactions between cancer cells and immune cells within a reconstructed TME. These assays provide critical insights into immune cell recruitment and cytotoxicity, key to understanding bispecific antibody mechanisms of action. Co-culture systems offer a high-throughput, controlled environment for testing bispecific antibodies, maintaining tumor heterogeneity and complexity, making them ideal for pre-clinical interrogation of different strategies to address the complexities of bispecific antibody research. In the hematologic malignancies field, leveraging primary, never-passaged, well-characterized patient material allows the evaluation of bispecific antibody efficacy in a clinically relevant ex vivo system where immune cell viability and function are retained. Conclusion The complexity of bispecific antibody mechanisms needs advanced preclinical platforms that accurately replicate human tumor biology, such as PDX in humanized in vivo hosts, organoids-based co-culture assays, and hematological platforms supporting immune cells, offering unparalleled tools for translational research. By integrating these platforms into bispecific antibody development, researchers can gain deeper insight into mechanisms of action, better predict therapeutic outcomes, and ultimately enhance clinical success. The future of bispecific, trispecific, and even pentaspecific antibody therapies and the combination with checkpoint inhibitors hinges on the optimal selection and execution of specific translational strategies leveraging these innovative preclinical models.
Advances in BCMA-Targeting Therapies: Takeaways from ASH2024
This year’s ASH conference saw a significant focus on multiple myeloma. Arcellx's Gilead-partnered CAR-T therapy, anitocabtagene autoleucel was a major highlight. At a follow-up of 34 months, a single anito-cel infusion delivered early, deep, and long-lasting responses in patients with heavily pretreated relapsed or refractory multiple myeloma (RRMM). Remarkably, the complete response rate reached 79%. Importantly, the treatment demonstrated a manageable safety profile.[1, 2] However, notable advancements in BCMA-directed multiple myeloma therapies extend beyond CAR-T, with a focus on antibody formats. An interesting update concerned ABBV-383, a T-cell engager developed by AbbVie following its acquisition of TeneoOne. Recent data highlight promising outcomes for ABBV-383 in combination with Darzalex and dexamethasone, yielding an overall response rate (ORR) of 70%, which rises to 82% at higher doses. This development builds on AbbVie's earlier report of a 57% ORR from ABBV-383 monotherapy trials and underscores AbbVie's commitment to this area.[3, 4, 5] Initial findings on EMB-06, a T-cell engager developed by EpiMab and subsequently licensed to Vignette Bio, were also presented [6]. Complementing these discussions of BCMA-targeted therapies, ASH also showcased updates on Bristol Myers Squibb's pipeline. This includes its immunomodulatory cereblon E3 ligase modulators (celmods), such as mezigdomide, golcadomide, and iberdomide, aimed at extending the legacy of its Celgene-derived multiple myeloma treatments. [7, 8, 9, 10, 11] Beyond BCMA, BMS's anti-GPRC5D CAR-T candidate, BMS-986393, has demonstrated significant potential, with Phase 1 trials reporting an impressive 91% ORR among evaluable patients treated at the go-forward dose. These results have encouraged the initiation of the Phase 3 Quintessential-2 trial, scheduled to begin in early 2024. [12, 13] This year’s ASH also explored the management of precancerous conditions like smoldering myeloma, a primarily asymptomatic precursor to active multiple myeloma. Discussions focused on whether aggressive treatments, such as CAR-T therapy, are appropriate for managing these cases. The therapy's risks are highlighted by a study of Carvykti showing severe neutropenia in all patients and liver enzyme elevations in a subset [14]. Johnson & Johnson and Genmab’s Darzalex has shown promise in treating smoldering myeloma, with significantly improved progression-free survival compared to active monitoring (hazard ratio 0.49, p<0.0001) [15, 16]. Sanofi’s Sarclisa, also targeting this condition, is progressing through trials [17]. ASH 2024 delivered critical updates on these advanced clinical trials and therapies. The intersection of novel treatment modalities and evolving clinical strategies ensured the central role of this year's conference to the ongoing innovation in multiple myeloma and related indications. Champions Oncology offers a cohort of patient-derived primary multiple myeloma models for use in our ex vivo Hematological Vitroscreen. These extremely rare and well-annotated models can help advance your multiple myeloma research and accelerate your drug pipeline.
How to Develop Relevant Co-Culture Models of the Tumor Microenvironment
The Tumor Microenvironment: A Barrier to Effective Cancer Therapy The tumor microenvironment or TME is critical in shaping tumor progression, therapy resistance, and recurrence. The tumor microenvironment comprises stromal cells, immune cells, and signaling molecules that significantly impact how tumors respond to treatment. Immunosuppressive components of the tumor microenvironment, such as M2-polarized macrophages and activated cancer-associated fibroblasts (CAFs), foster resistance to therapies by creating an environment where immune responses are suppressed, and tumor survival is promoted. Key cytokines like IL-10 and TGF-β drive this immunosuppressive milieu, hindering the efficacy of immunotherapies and other anti-cancer treatments. Despite advancements, many cancer therapies fail in the clinical setting because the preclinical models used to evaluate them inadequately replicate the human tumor microenvironment. Addressing this issue is crucial for developing treatments that overcome tumor microenvironment-induced resistance and enhance clinical outcomes. The Limitations of Current Preclinical Models In Vivo Models: The Gold Standard, But Not Without Flaws In vivo models, particularly mouse models, have long been considered the gold standard for preclinical cancer research. However, they are not perfect, and it is important to know their limitations to navigate the field and select the right model for every application. While patient-derived xenografts (PDXs) maintain the genetic and histopathological features of human tumors and are considered an optimal model for clinical translatability of pre-clinical testing, the stromal compartment—including vasculature, immune cells, and fibroblasts—originates from the mouse host. This murine stroma fails to fully replicate the complexity and behavior of the human tumor microenvironment, limiting translational relevance. Humanized mouse models attempt to overcome some of these limitations by engrafting human immune cells into immunodeficient mice. While this approach provides valuable insights into immuno-oncology, it does not incorporate other critical human stromal components, such as CAFs and endothelial cells. This leaves a significant gap in accurately modeling the human tumor microenvironment, particularly when studying immunosuppressive mechanisms and therapy resistance. Simpler Models: Insufficient for Complex Therapeutic Mechanisms Monocultures of cancer cell lines, while failing to account for the intricate interactions between tumor cells and the surrounding tumor microenvironment, also lack tumor heterogeneity. The absence of stromal components in these models overlooks key factors influencing therapy mechanisms of action (MoA), particularly those targeting the tumor microenvironment. For example, therapies designed to modulate immune responses or disrupt fibroblast-mediated immune suppression are significantly affected by the presence and functionality of tumor microenvironment components. Consequently, these simplistic models often yield non-translatable results, leading to failure in clinical trials. The Tumor Microenvironment's Role in Resistance, Recurrence, and Progression The tumor microenvironment is a dynamic and interactive ecosystem that evolves with tumor progression. Specific elements of the tumor microenvironment, such as M2 macrophages and CAFs, actively promote resistance and recurrence by suppressing anti-tumor immunity and enhancing tumor survival. M2 Macrophages secrete IL-10, TGF-β, and VEGF, promoting immune evasion, angiogenesis, and tumor progression. CAFs can contribute to the desmoplastic reaction and produce extracellular matrix proteins, such as collagen, which act as physical barriers to therapy. They also release CXCL12 and TGF-β, further suppressing immune cell infiltration and activity. Therapies targeting these immunosuppressive mechanisms have shown promise in preclinical settings but often fail in clinical trials due to the lack of a human-relevant tumor microenvironment in the models used for their development. To address these challenges, advanced ex-vivo co-culture models that mimic the human tumor microenvironment are essential. These models integrate multiple human cellular components, including tumor cells, immune cells, fibroblasts, and endothelial cells, in ratios and conditions that closely resemble specific tumor types. Advantages of Ex Vivo Co-Culture Models By using human cells for all tumor microenvironment components, these models eliminate the cross-species differences inherent in mouse models. The proper characterization and ratios of human stromal cells enable accurate modeling of tumor microenvironment-driven mechanisms, such as immunosuppression and fibrosis. Real in-depth research is needed on the tumor microenvironment composition of different human tumors before starting to generate co-culture mimicking the tumor microenvironment. To provide better translational relevance, it is ideal to leverage Patient Derived Xenograft-organoids (PDX-O) a valuable source of tumor cells that retain the heterogeneity and genetic complexity of the original tumor providing data that more closely predict clinical outcomes, reducing the high failure rate of therapies in clinical trials. Ultimately, using PDX-Os combined with human stromal and immune components in co-culture models creates a system that mirrors the patient-specific tumor microenvironment, enabling the evaluation of therapies in a clinically relevant context. Applications of Ex Vivo Tumor Microenvironment Models: CAR-T Cell Therapy in Immunosuppressive Tumor Microenvironment CAR-T cell therapy, a breakthrough in cancer immunotherapy, often struggles in solid tumors due to the immunosuppressive TME. M2 macrophages and CAFs inhibit CAR-T cell infiltration and activity through the secretion of TGF-β and CXCL12. Recent studies have demonstrated that anti-TGF-β antibodies or CXCR4 inhibitors can restore CAR-T cell functionality in ex-vivo co-culture models, highlighting the importance of these models for optimizing CAR-T therapeutics targeting CAFs [1]. Similarly, antifibrotic agents, such as inhibitors of lysyl oxidase (LOX) or TGF-β signaling, aim to disrupt the fibrotic stroma created by CAFs. Using ex vivo tumor microenvironment models, researchers have shown that these agents enhance the penetration and efficacy of chemotherapies and immunotherapies [2]. This highlights the key role these models can play in evaluating combination strategies targeting both the tumor and its microenvironment. Developing Strategies to Mimic the Human Tumor Microenvironment Creating robust ex-vivo tumor microenvironment models requires careful selection and characterization of human cells. Strategies include the selection of the right tumor microenvironment cells from the right origin, using the best E:T ratio between immune cells and patient-derived tumor cells, and defining optimal ratios of immune cells, fibroblasts, and endothelial cells to replicate a model-specific tumor microenvironment. In addition, functional validation is fundamental in ensuring that all cellular components behave as they do in vivo, including cytokine secretion and immune cell polarization. These considerations are critical for generating data that can effectively guide clinical decision-making. Bridging the Gap to the Clinic The development of human-relevant ex vivo co-culture models represents a significant step forward in cancer research. By faithfully replicating the human tumor microenvironment, these models provide a powerful platform for studying therapy resistance, optimizing treatment strategies, and improving the clinical translatability of preclinical findings. Ultimately, this approach aligns with the goal of delivering better outcomes for cancer patients.
Bad Blood: Modeling Biologically Relevant Blood Cancer Studies
In the vast and intricate world of cancer research, blood cancers—such as leukemia, lymphoma, and myeloma—pose unique challenges. Unlike solid tumors, these malignancies originate in the bone marrow and affect the production and function of blood cells. For scientists, understanding these diseases is crucial for developing effective treatments and enhancing patient outcomes. The quest to understand blood cancer has driven scientists to develop innovative modeling techniques that mimic the disease's progression and response to treatments. Among these, ex vivo and in vivo models stand out as vital tools, each offering distinct insights and challenges. This blog post will explore the intricacies of these blood cancer modeling approaches, highlighting their advantages, limitations, and relevance in modern-day blood cancer research. Understanding Ex Vivo and In Vivo Modeling in Blood Cancer Studies To fully appreciate the nuances of blood cancer research, one must first grasp the fundamental differences between ex vivo and in vivo modeling. Ex vivo models refer to experimental setups where peripheral blood cells or bone marrow that contain the leukemic cells are taken from an organism and studied outside of their natural environment, in a controlled laboratory setting. This allows researchers to examine cellular behaviors and responses with precision, free from the complexities of a living organism. In contrast, in vivo models of blood cancer involve studying the disease within a living organism, typically using animal models like mice. This approach provides a more holistic view of how a disease behaves in a complex biological system, considering factors such as immune responses and interactions with other tissues. While both models are invaluable for blood cancer research, their applications and insights can vary significantly depending on the research question at hand. Advantages and Limitations of Ex Vivo Models The use of ex vivo models in blood cancer research is favored by the access to tumor cells from the blood. This approach allows for a direct detailed examination of cellular processes, enabling researchers to manipulate and observe how blood cancer cells react to specific treatments or conditions soon after they are extracted from the patient. This level of control is essential for understanding how certain therapies affect blood cancer cell survival and proliferation. As such, ex vivo models can provide rapid results and researchers can quickly gather data and adjust their experiments accordingly. This accelerates the pace of discovery and innovation in the blood cancer field. Primary Blood Cancer Models vs Immortalized Cell Lines One significant advantage of primary blood cancer models over cell lines is their ability to more accurately reflect the genetic and phenotypic diversity of actual patient tumors. Cell lines, although useful, undergo genetic drift and become less representative of the original tumor's complexity. In contrast, primary models of blood cancer, derived directly from patients' samples, retain the heterogeneity and specific characteristics of the patient's disease. This fidelity ensures more reliable insights into the tumor's behavior and response to therapies, which is crucial for developing personalized treatment strategies and improving clinical outcomes in blood cancer research. Co-Cultures in Primary Blood Cancer Models Co-cultures in primary blood cancer models provide an advanced method to closely simulate the tumor microenvironment by cultivating blood cancer cells alongside other relevant cell types, such as stromal or immune cells. This technique enriches the blood cancer model's complexity, shedding light on critical cellular interactions and signaling pathways that drive blood cancer progression and resistance to treatments. By incorporating multiple cell types, co-cultures facilitate exploration of the tumor cell clonality and dynamic interaction with the surrounding microenvironment by flow cytometry and/or high-content imaging within a more physiologically relevant context. Consequently, they enhance the accuracy of predictions related to therapeutic responses and enable the development of more targeted and effective treatment strategies for blood cancers. Limitations of Ex Vivo Models Ex vivo models of blood cancer are not without their limitations. The primary challenge lies in their inability to replicate the complex interactions that occur in a living organism. Factors such as immune responses, microenvironmental influences, and systemic effects are often absent in ex vivo setups, potentially leading to results that may not fully translate to in vivo scenarios. Advantages and Limitations of In Vivo Models In vivo models bring a different set of strengths to blood cancer research. Their greatest advantage is the ability to study blood cancer within the context of an entire living system. This provides insights into how blood cancer interacts with the host's immune system, how treatments affect overall health, and how the disease may evolve over time. Through in vivo blood cancer studies, researchers can observe the effects of a treatment on both the tumor and the host. This holistic view is crucial for understanding not only the efficacy of a therapy but also its potential side effects and long-term consequences. Despite these benefits, in vivo models of blood cancer have their own set of challenges. They can be time-consuming and costly, requiring significant resources to maintain and execute. Finally, there is always the risk that findings in animal models of blood cancer may not perfectly translate to human patients. Challenges of Modeling Blood Cancer In Vivo Patient-derived xenograft (PDX) models can be generated for some blood cancer indications. This approach involves implanting patient tumor cells into immunocompromised mice and passaging the tumor into a series of mice to establish a stable model. Although these blood cancer models are a better representation of the clinical disease compared to cell lines, due to passaging, PDX models from multiclonal blood cancer such as AML would not retain the cellular and molecular heterogeneity typical of the patient’s disease, limiting their clinical relevance. Instead, primary patient-derived models of blood cancer created by implanting patient tumor cells into immunocompromised mice for in vivo studies not only preserve the genetic characteristics of the original tumor but also, and most importantly, retain the heterogenic nature of the patient’s disease, therefore providing a closer representation of blood cancer like AML in patients. However, they can be difficult to develop and maintain, limiting their widespread use. The Importance of Diverse Modeling Approaches in Blood Cancer Research In the quest to cure blood cancer, no single modeling approach offers all the answers. Both ex vivo and in vivo models of blood cancer have their place in the research ecosystem, each contributing valuable insights to our understanding of these complex diseases. For scientists, the key lies in leveraging the strengths of each approach and exploring new technologies that bridge the gap between precision and biological relevance in blood cancer research. By doing so, we can continue to push the boundaries of what is possible in blood cancer research, ultimately improving the lives of patients worldwide. Champions Oncology has assembled a comprehensive collection of platforms encompassing a diversity of blood cancer types, including AML, B-ALL, T-ALL, CLL, DLBCL, MCL, MDS, and MM, directly from primary patient samples. This living bank of primary tumors empowers our clients to evaluate the efficacy of innovative therapeutic strategies with remarkable precision both in vivo and ex vivo. By encapsulating the intricate biology of blood cancer and mirroring the considerable heterogeneity inherent in patient populations, our platform is at the forefront of facilitating a rapid transition from bench to bedside.
Primary Blood Cancer Models: Getting Blood from a Stone
In the intricate maze of biomedical research, the quest for accuracy and relevance often leads to one pivotal question - which model systems offer the most reliable insights? The choice of a reliable system becomes even more mandatory for hematologic malignances, and in particular Acute Myeloid Leukemia (AML), that are inherently characterized by a high degree of heterogeneity. Among all the options, primary blood cancer models stand out as high-fidelity systems. These models, rooted in the direct application of human samples, are redefining how researchers approach complex biological questions. For those, scientists focused on pharmaceutical development in oncology, understanding the nuances of these models could unlock new pathways in their investigations. Why Primary Blood Cancer Models Are Gaining Ground The use of primary blood cancer models is critical for scientists to be able to mirror clinical outcomes, however, the field is still impacted by the access to only poor-quality models. Understanding why primary blood cancer models are increasingly preferred over cell lines and, in some cases, over traditional patient-derived xenograft (PDX) models is imperative. Specifically, for some hematological malignancies such as AML, it has been shown that there is an important loss of disease multiclonality at early passages [1]. Primary AML Models’ Edge Over PDX Models When it comes to AML, primary models offer several distinct advantages over serially passaged PDX models. First, they provide a closer genetic match to the human condition, enabling more precise interpretations of how tumors behave in vivo. Additionally, the primary models maintain the original AML heterogeneity, providing valuable insights into the mechanisms driving tumor progression and drug response. This fidelity is crucial for researchers aiming to unravel the complexities of AML biology. Unlike serially passaged AML PDX models, which can lose critical human-specific characteristics over time due to adaptation in a non-human host and undergo clonal selection through passaging, primary AML models maintain cellular integrity and relevance. A Step Towards Personalized Medicine By mirroring human physiological conditions more accurately, primary blood cancer models facilitate the development of personalized medicine. Researchers can test how individual patients might respond to specific treatments, paving the way for tailored therapeutic strategies. This approach may not only improve patient outcomes but also enhance the efficiency of clinical trials by identifying the most promising candidates early in the process. Biopharma Development on the Horizon For biopharma companies, primary heme models present opportunities for innovation. These models allow scientists to identify potential drug candidates more rapidly and with greater accuracy, minimizing the risk of late-stage clinical trial failures. By incorporating primary blood cancer models into the drug development pipeline, biopharma companies can streamline their processes, reduce costs, and ultimately bring life-saving therapies to market faster. A New Era for Translational Research Translational research aims to bridge the gap between laboratory findings and clinical applications. Primary blood cancer models serve as a catalyst in this process, enabling researchers to translate basic scientific discoveries into therapeutic interventions more efficiently. Their ability to mimic human physiology closely ensures that findings in the lab are more likely to be relevant to patient care, enhancing the overall impact of research efforts. Practical Tips for Incorporating Primary Blood Cancer Models While the benefits of primary blood cancer models are clear, integrating them into research methodologies requires careful planning and execution. Here are some practical tips for researchers looking to harness the power of these models. Building a Robust Infrastructure Establishing a successful primary blood cancer model system begins with creating a robust infrastructure. This includes acquiring high-quality primary human blood samples, ensuring proper storage and handling protocols, and investing in the necessary equipment and technology. Collaborative partnerships with hospitals and biobanks can facilitate access to diverse tissue samples, enhancing the diversity and applicability of the research. Working with an expert provider that has access to a deep clinical network can solve both the sample procuring and technical complexity issues. Champions Oncology offers the largest bank of engraftable primary blood cancer models. Our continued effort in sourcing the most clinically relevant tumors and our proved expertise in hematological tumor studies make us the best partner to help you advance your blood cancer pipeline. Addressing Common Challenges and Misconceptions Despite their potential, primary blood cancer models come with their own set of challenges and misconceptions. It is crucial for researchers to be aware of these and adopt strategies to overcome them. Overcoming Technical Limitations One common challenge is the technical complexity involved in establishing and maintaining primary blood cancer models. Researchers must be diligent in optimizing culture conditions, monitoring cell viability, and ensuring the reproducibility of results. Regular quality checks and standardization of protocols can mitigate these challenges and improve the reliability of the models. Researchers should also remain open to exploring new technologies and methodologies that can enhance the performance of primary blood cancer models. Continuous innovation and adaptation are essential to address evolving research needs and maximize the potential of these models. Debunking Misconceptions There are several misconceptions surrounding the use of primary blood cancer models, particularly regarding their cost and scalability. While initial investments may be required, the long-term benefits of these models often outweigh the costs. Their ability to provide more accurate and relevant insights can lead to more successful research outcomes and, ultimately, cost savings. Another misconception is that primary blood cancer models are only suitable for specific research areas. In reality, their versatility makes them applicable across a wide range of biomedical fields, from cancer research to regenerative medicine. Researchers should explore the diverse applications of these models and consider incorporating them into their own research endeavors. In conclusion, primary blood cancer models represent a significant step forward in biomedical research. Their ability to provide accurate, relevant insights makes them an invaluable tool for researchers across a wide range of fields. By adopting these models, researchers can enhance their research outcomes, drive innovation, and ultimately improve patient care.
It’s All in the Blood: Choosing Clinically Relevant Blood Cancer Models for Research
Acute Myeloid Leukemia (AML) presents a remarkable challenge in oncology, characterized by the rapid growth of abnormal white blood cells. Developing clinically relevant research models is crucial for scientists to understand and treat AML effectively. These models can simulate the disease's complexity, providing insights into its progression and aiding in the development of targeted therapies. The Importance of Heterogeneity in AML One of the most significant hurdles in AML research is its heterogeneity. AML is not a homogeneous disease but a collection of diverse subtypes, each with its own genetic and molecular characteristics. Each patient's tumor is composed of several different malignant myeloid blast cell subpopulations that make each tumor unique. This diversity impacts clinical outcomes and treatment responses, making it essential for researchers to use models that accurately reflect this heterogeneity. By acknowledging and incorporating this variability, researchers can better predict patient responses to new treatments. (Read our blog "A Needle in a Haystack: Finding Rare AML Populations by Flow Cytometry") Exploring AML Model Options There are several AML models available, each offering unique insights and benefits: Cell Line Models Cell line models are often the first step in AML research. These models allow researchers to study specific cellular processes in a controlled environment. However, they do not capture the complexity of the disease due to their homogeneous nature. Despite this limitation, cell lines can be useful for initial screenings and mechanistic studies. Patient-derived xenografts (PDX) PDX models involve implanting patient tumor cells into immunocompromised mice and passaging the tumor into a series of mice to establish a stable model. Although these models are a better representation of the clinical disease compared to cell lines, due to passaging, PDX models do not retain the cellular and molecular heterogeneity typical of AML, limiting their clinical relevance. Primary patient-derived models Primary patient-derived models are created by culturing primary AML cells from patient samples for ex vivo use or implanting patient tumor cells into immunocompromised mice for in vivo studies. These models not only preserve the genetic characteristics of the original tumor but also, most importantly, retain the heterogenic nature of the patient’s disease, therefore providing a closer representation of AML in patients. This approach also preserves the tumor's microenvironment, allowing researchers to study how it interacts with treatments. However, they can be difficult to develop and maintain, limiting their widespread use. (Learn about our ex vivo hematological model offering) Champions’ Medical Affair team continuously procures primary AML samples representative of the heterogeneity of the patient population, and our expert team develops and validates their growth in our experimental settings. We offer a large bank of primary, never passaged, AML models for ex vivo and in vivo use. Our oncology experts can help you achieve your research objectives from model selection and study planning to data analysis and result interpretation. (Explore our in vivo hematological model offering) Selecting the Right Type of AML Model Choosing the appropriate AML model depends on the specific research objectives. Researchers should as always try to match the research question with the complexity of the model, and the available resources. For instance, cell line models may suffice for preliminary studies, while patient-derived models are preferable for testing targeted therapies. Collaborations with experienced organizations, like Champions Oncology, can provide valuable insights and access to tailored experimental solutions. (Contact us) The Role of Clinical Annotations Incorporating clinical annotations into AML model characterization is crucial to select the right models for replicating patient scenarios accurately. Champions primary AML models are deeply characterized and extensively annotated. These annotations provide context, linking laboratory findings to real-world patient data. By integrating molecular characteristics with clinical information such as treatment history and patient outcome, researchers can enhance the relevance and predictive power of the results obtained. This approach bridges the gap between experimental research and clinical application. (Dive into Lumin, our extensive multi-omics and multimodal database at lumin.bio) The ongoing evolution of models and technologies promises a brighter future for AML research, ultimately bringing us closer to effective treatments and cures. The choice of clinically relevant AML models is vital for advancing oncology research in this field. By recapitulating the disease heterogeneity and incorporating clinical annotations, Champions’ AML models from primary patient samples help researchers drive meaningful discoveries that will eventually improve patient outcomes. By leveraging the expertise of leading organizations like Champions Oncology, you can access the tools and insights needed to elevate your research and make a lasting impact in the fight against AML.
PDX Models: Masters of Diversity
Cancer research is constantly evolving, seeking new methods to better understand and treat this complex disease. Among the most groundbreaking developments in this field are patient-derived xenograft (PDX) models. These models have revolutionized our approach to studying tumors by preserving the heterogeneity and molecular characteristics of a patient's tumor. This blog post aims to provide an in-depth look at the heterogeneity of PDX models and why it’s important to have diversity in your preclinical in vivo studies. The Importance of PDX Models Unlike traditional cell line-based models, PDX models retain the genetic diversity and molecular complexity of the original tumor. PDX models offer researchers a closer approximation of how human tumors behave in vivo, with different cell populations responding differently to therapies. They provide a dynamic and clinically relevant platform for studying tumor growth, metastasis, and response to treatment, making them a powerful tool in the fight against cancer. (Read our blog "The Ultimate Guide to Designing a Mouse Clinical Trial and Data Analysis") Models pretreated with targeted therapies are ideal to test next-generation agents. For example, when developing a drug that aims to overcome resistance to the standard of care therapy in a given tumor type, choosing models pretreated with that standard of care agent for our efficacy study will reveal true therapeutic effects in the target patient population. (Read our blog "Accelerating Innovation & Drug Development with Pre-treated PDX Models") Tumor Heterogeneity of PDX models is important for translational oncology studies One of the most significant advantages of PDX models is their ability to preserve tumor heterogeneity. Tumors are not uniform; they consist of various cell types with different genetic mutations and behaviors. On the contrary, traditional cell lines are developed in vitro by clonal selection, leading to a less accurate representation of the human disease.[1] PDX models also retain the molecular characteristics of the original tumor. This includes genetic mutations, epigenetic modifications, and gene expression patterns. By maintaining these features, PDX models provide a more accurate representation of the human tumor genetic background, which is crucial for developing effective therapies.[1] Advancing Cancer Research with PDX Models The ability of PDX models to preserve tumor heterogeneity and molecular characteristics has significantly advanced cancer research. Researchers can study the behavior of different tumor subpopulations, identify potential targets for therapy, and evaluate the efficacy of new treatments in a more realistic setting. PDX models have heterogeneous biomarker expression. The figure below, for example, shows the expression of KRAS in Champions’ PDX models varying across and within tumor types. Selecting PDX models with a higher KRAS expression will be paramount to successfully test KRAS inhibitors and could provide more clinically relevant results. Nonetheless, a PDX model with elevated KRAS expression will still present with heterogeneous expression across the different cell clones within the tumor, a setting that realistically reproduces the human tumor heterogeneous cell composition. In this setting, a KRAS inhibitor might only be partially effective at killing the tumor because cell clones with lower KRAS expression within the tumor might not be affected, resulting in lower overall therapeutic efficacy. Similarly, in the clinic, we could observe a partial response followed by tumor progression, due to the selection of the low KRAS-expressing cell clone within the tumor. For this reason, studies that use PDX models instead of cell line-derived models will produce more reliable and clinically relevant results. KRAS gene expression across Champions' PDX models of different tumor types. Patient-derived xenograft models have revolutionized cancer research by providing a more accurate representation of human tumors. Their ability to preserve tumor heterogeneity and molecular characteristics makes them an invaluable tool for studying cancer biology, testing new therapies, and personalizing treatment plans. By continuing to develop and refine these PDX models, researchers can accelerate the discovery of new treatments and bring them to patients faster. Champions Oncology has over 1500 clinically relevant PDX models that can accelerate your oncology research program. Our models have broad clinical annotation and have been deeply characterized using NGS (WES & RNA-seq), proteomic, and phospho-proteomic datasets to interrogate the heterogeneity of the tumor. To learn more about our PDX tumor models, access Lumin Analytics.
A Step-by-Step Guide to Design a 3D Tumor Model Co-Culture Study
Designing a 3D tumor model co-culture study in oncology research is a meticulous process that demands precision and expertise. However, when executed correctly, these studies provide invaluable insights into tumor-immune microenvironment interactions, potentially leading to groundbreaking therapeutic advancements. This guide outlines essential steps to design a robust 3D tumor model co-culture study, ensuring you capture high-quality data and actionable insights. 1. Select 3D Tumor Models and Validate Target Expression Mechanism of Action: Begin by selecting cancer type, 3D tumor models, and co-culture strategies that are appropriate for the mechanism of action of your therapeutic agent. This involves understanding the biological pathways and processes that your agent targets, ensuring that the 3D tumor models accurately reflect these conditions. Target Expression-Driven Model Selection: Select 3D tumor models based on target expression to best set up your ex-vivo co-culture assay. Leveraging a fully characterized bank, such as Champions’ tumor model bank, is essential for successful model selection. Access to transcriptomic data, for example, allows to rank order the 3D tumor models based on different gene expression profiles, making model selection easy and effective. (Champions’ entire database is accessible for model selection in Lumin) Validation Techniques: Validate target expression using PCR, western blot, or flow cytometry techniques. PCR can help detect and quantify specific nucleic acid sequences, western blotting can confirm protein expression levels, and flow cytometry can analyze the physical and chemical characteristics of cells, ensuring the presence and activity of your targets. This step is crucial for confirming that your targets are being correctly expressed and are functional within your chosen models. Read our blog "Using 3D Ex Vivo Tumor Models for Oncology Research: An Expert Guide" to learn more about the advantages of using tumor organoids in cancer research. 2. Select Multiple Immune Cell Donors to Account for Donor-to-Donor Variability In 3D tumor model co-culture studies, accounting for donor variability is crucial for the robustness of your findings. Selecting immune cell donors from a diverse pool minimizes the risk of skewed results that could arise from individual donor-specific traits. Ensure that immune cells are sourced from multiple donors to account for genetic, epigenetic, and environmental variations. This approach enhances the generalizability and reliability of your study outcomes. Additionally, thorough donor screening and characterization using flow cytometry can further refine your selection process, providing a comprehensive understanding of each donor’s immune profile. Autologous Immune Cells: Autologous immune cells, sourced from the same patient as the 3D tumor model, offer several significant benefits. One of the primary advantages is the enhanced compatibility, as these cells are naturally tailored to interact with the patient’s specific tumor microenvironment. This compatibility can lead to more accurate reflections of in vivo interactions, allowing for better predictive modeling of therapeutic responses. Additionally, using autologous cells can help mitigate the risk of immune rejection, further promoting a more sustained interaction in the 3D tumor model co-culture setting. Despite their advantages, autologous immune cells can present challenges, including donor variability and limited availability. The unique immunological characteristics of each patient can create substantial variability in the efficacy and functionality of the immune cells, leading to inconsistencies in reproducibility. Moreover, some patients may not have accessible immune cells, which can hinder the study's progress and breadth. Allogeneic Immune Cells: On the other hand, allogeneic immune cells, derived from different donors, offer advantages in terms of standardization and availability. These cells can be sourced from healthy donors or patients with known, consistent immune profiles, allowing for high-throughput studies and reproducible results. This standardization can enhance the robustness of the findings, as variations between immune responses can be systematically controlled and compared. However, allogeneic immune cells come with their own set of challenges. The primary concern is the potential for immune rejection, as these cells may not be recognized as 'self' by the 3D tumor model or surrounding microenvironment. This disconnect can influence the dynamics of tumor-immune interactions and may not accurately represent the patient-specific responses, ultimately resulting in less applicable data for therapeutic strategies. Furthermore, the genetic and environmental differences between donors can lead to variability in immune responses, complicating the interpretation of results. Champions supports your ex vivo immuno-oncology program offering both autologous and allogeneic immune cells for 3D tumor model co-culture assays. (In our webinar "Autologous Co-culture Models to Humanized Mouse Models: Navigating IO Models" our experts deep dive into the intricate world of immuno-oncology models to help you identify the best model for your study) 3. Assess Tumor-to-Immune Cell Ratio Achieving the right composition and cell population balance when replicating the tumor microenvironment (TME) ex-vivo is crucial in designing a 3D tumor model co-culture system. One of the strengths of ex-vivo 3D tumor model co-cultures is the ability to customize different parameters to recreate the TME of interest, providing a physiologically relevant platform to predict clinical outcomes. Origin of the Cellular Component and Phenotype: It is very important to source each cell population from the right tissue. When introducing a new cell type within the 3D tumor model co-culture system, it is fundamental to make sure each cellular component is functional and expressing the correct differentiation markers. In some cases, it is necessary to differentiate progenitor cells into the desired cell type before adding them to the 3D tumor model co-culture. In other experimental settings, activating the immune cells to induce specific target expression is crucial to resemble diseased TME. Autologous vs Allogeneic Population Assessment: Verify the necessary stroma and immune cell populations using advanced techniques such as flow cytometry and expression profiling. Flow cytometry allows for the detailed analysis of cell populations, including their size, granularity, and protein expression. Expression profiling further assesses the expression of the correct differentiation markers by stroma cells and the target expression and activation status of the immune cells, ensuring they are primed and ready to attack the tumor cells effectively. Cell Ratios: It's essential to determine the optimal ratio of tumor cells to immune cells to ensure the immune cells can effectively target and eliminate the tumor cells. This involves careful calculation and adjustment based on the specific characteristics of the tumor, TME, and immune cells being used. By carefully balancing cell ratios, type, and status, and thoroughly assessing cell populations, we can control the 3D tumor model co-culture system's ability to evaluate cancer immunotherapies and improve clinical translatability. 4. Identify the Right Assay Controls Controls are pivotal for obtaining reliable results and ensuring the validity of an assay. There are several types of controls to consider: Mechanism-Based Controls: These are crucial for the assay as they should be selected based on the specific mechanism of action of the therapeutic agent being tested. The correct set of controls will allow a better understanding of how the therapeutic agent in preclinical stage of development will behave in the given biological context. In particular, this type of control is essential to interpret mechanistic findings in ex-vivo 3D tumor model co-culture systems. Validation: It is essential to implement these controls consistently throughout the entire assay process. This step ensures that the results are accurate and reproducible, minimizing false positive and negative readouts which is fundamental for drawing meaningful conclusions from the data. In addition, using these controls, researchers can ensure the ex-vivo 3D tumor model co-culture system is responding as expected to a given perturbation and it can provide meaningful results in the specific biological context of interest. By carefully choosing and applying these controls, researchers can significantly improve the reliability and validity of their experimental results. 5. Choose the Best Readout for Tumor Killing Selecting the right readout method is critical for obtaining meaningful data: Imaging: Utilize advanced imaging techniques for visualizing 3D tumor model cell interactions and capturing detailed images that provide insights into cellular behavior and morphology. The high throughput confocal approach provides meaningful images to have a glance into the 3D tumor model co-culture system. Moreover, the image quantification pipeline allows for quantitative data extraction through 3D image reconstruction. Beyond tumor killing, immune cell infiltration into the 3D tumor model is another parameter that can be evaluated through an accurate image analysis. Flow Cytometry: Assess different phenotypes within the diverse cell populations composing the co-culture, viability (which provides a direct tumor-killing measure), and specific target expression using flow cytometry. This technique allows for precise quantification and analysis down to the single cell level, dissecting the heterogeneous populations within the 3D tumor model co-culture system. Luciferase: Employ luciferase-based assays for fast, quantifiable, and direct measurement of cancer cell killing. This endpoint requires the use of luciferase-tagged cells, enabling the measurement of gene expression, cellular activity, and other biological processes through luminescence. (Watch our on-demand webinar "Poster QuickTake: Bioluminescent 3D Tumors with Immune Cell Co-culture for HTS" to learn more about bioluminescence tagged 3D tumor models for co-culture assays) 6. Use Immune Cell Infiltration as an Additional Readout Immune cell infiltration offers deeper insights into the body's response to cancer: Infiltration Analysis: Track immune cell movement into the tumor microenvironment to gauge the effectiveness of your therapeutic agent. This analysis helps in understanding the behavior of immune cells and their interaction with 3D tumor model cells, which is crucial for developing more effective treatments and improving patient outcomes. The 3D organization of the cells in different compartments is important in this kind of analysis to decide which parameter is best to use in evaluating cellular mobility. 7. Assess Immune and Stroma Cell Status Post-Treatment Post-treatment analysis of the 3D tumor model co-culture system helps interpret therapeutic effects by providing detailed insights into cellular and molecular responses. Here are two key techniques: Flow Cytometry: Use this technique to analyze immune and stroma cell phenotypes and statuses. It allows for the identification and quantification of various cell types within a sample, offering a comprehensive overview of changes in the cellular landscape following treatment. Cytokine Analysis: Measure cytokine levels to understand the immune response and stroma cell dynamics post-treatment. By quantifying specific cytokines, researchers can infer the activation and regulation of different immune pathways, shedding light on the effectiveness and impact of the therapeutic intervention. Designing a 3D tumor model co-culture study is a complex yet rewarding endeavor that can yield significant insights into tumor biology and treatment efficacy. By following these steps, you can ensure your study is thorough, accurate, and highly informative. Ready to take your 3D tumor model co-culture studies to the next level? Partner with Champions Oncology for unrivaled expertise and cutting-edge resources to propel your research forward. We tailor our services to meet your specific research needs. Contact us today to learn more!

How to use metadata as your model selection GPS
In the complex landscape of oncology research and drug development, precision is paramount. The ability to select appropriate models can significantly impact the success and validity of your research. This is where metadata steps in as your reliable guide for model selection. Understanding Metadata in Research Metadata, essentially data about data, plays a crucial role in organizing, describing, and finding datasets. In the context of oncology research, metadata encompasses all the details about experiments, datasets, and results. This includes information on sample types, experimental conditions, data collection methods, and software used. By providing a structured summary of data, metadata helps streamline the process of data retrieval and utilization, for model selection and complex data analysis. Metadata serves as a roadmap, guiding researchers to the datasets they need, while also providing context and ensuring that data is used appropriately. It’s an essential tool for maintaining consistency and transparency in research. The better structured and more detailed the metadata, the easier it is to manage and interpret large datasets, which are common in oncology. In oncology research, metadata might include details such as the type of tissue samples collected, the sequencing methods used, and the specific conditions under which experiments were conducted. This ensures that datasets remain comprehensible and reusable, even years after the initial research was completed. The Importance of Accurate Metadata in Model Selection Accurate metadata is the backbone of effective model selection. It provides the necessary context to understand datasets fully, ensuring that models are selected based on relevant and precise information. This is particularly important in oncology research, where the accuracy of findings can have significant implications for patient outcomes. Champions Oncology’s entire database can be explored accessing Lumin, our proprietary integrated software solution that harnesses the power of precisely curated oncology data, computational science, and bioinformatics to help you make data-driven decisions in model selection and data interpretation. Within Lumin you can: visualize proprietary genomic, proteomic, and pharmacological profiles from Champions’ patient-derived preclinical models; generate analyses on unique multi-omic datasets assembled from over 12,000 patients, including thousands of clinical treatment responses not available in public databases; interrogate experimental data and accelerate your oncology research programs. Enhancing Model Selection With accurate metadata, researchers can filter through vast amounts of data to find the most relevant datasets for their specific needs. For instance, if you're looking for data on a particular cancer type and drug target, well curated metadata can help you quickly identify and access the relevant datasets, saving time and resources. Scientists planning to study the efficacy of a novel targeted therapy developed to treat tumors of a specific type that are resistant to the standard of care for that tumor type could use metadata to select the best set of PDX models to use in their study. Within Lumin, they could filter our bank of tumor models by tumor type first, and then, by drug response profile. In the example below, we selected all NSCLC PDX models derived from patients who were pre-treated with osimertinib and that did not respond to osimertinib treatment in vivo. This set of models would well represent the target patient population for our drug. Additionally, scientists could further select models based on gene mutation and RNA and/or protein expression levels of their agent’s target. A deeper look into the tumor model metadata, leveraging data analysis and visualization tools, can help fine-tuning model selection further improving the quality of the study results. Deep characterization and extensive clinical annotation of patient-derived tumor models, allows for a more precise model selection, which translates into a well-designed mouse clinical trial that closely reproduces the clinical setting and patient target population, and ultimately leads to more accurate results and a faster track to clinical experimentation. Best Practices in Structuring Metadata To harness the full potential of metadata, it's essential to follow best practices in its structuring and management. Here are some key guidelines for oncology researchers. Standardized Formats Using standardized formats for metadata ensures consistency and compatibility across different datasets and platforms. This makes it easier to integrate and compare data from various sources, enhancing the overall quality and reliability of your research. Detailed Descriptions Detailed descriptions of the datasets, including information on the origin of the data, the methods used for collection and analysis, and any relevant environmental or experimental conditions are very important. The more detailed your metadata, the more useful it will be for model selection and to bridge with future research. Regular Updates Keeping metadata up-to-date to reflect any changes or new findings is paramount. Regular updates ensure that data remains relevant and accurate, supporting the ongoing use and application of any research findings. Future Trends in Metadata for Research and Biotech The landscape of metadata is continually evolving, driven by advances in technology and increasing demands for data accuracy and transparency. Here are some future trends that are likely to shape the use of metadata in oncology research. AI and Machine Learning Artificial intelligence (AI) and machine learning are set to revolutionize the way we manage and utilize metadata. These technologies can automate the process of metadata generation and analysis, making it faster and more efficient. For example, AI algorithms can analyze large datasets to identify patterns and correlations that might be missed by human researchers. Integration with Big Data As the volume of data in oncology research continues to grow, there will be an increasing need for robust metadata systems that can handle big data. Integrating metadata with big data platforms will enhance the ability to manage, analyze, and interpret vast amounts of information, leading to more accurate and comprehensive research outcomes. Metadata is an invaluable tool for oncology researchers, providing the context and structure needed to manage complex datasets effectively. By following best practices in model selection, you can enhance the accuracy and efficiency of your research, leading to better outcomes and discoveries. The incorporation of metadata into your research processes not only supports better model selection and data integrity but also fosters collaboration and innovation. Ready to advance your oncology pipeline with faster model selection and AI-powered data analytics & visualizations? Sign up for our free trial of Lumin and discover how we can help you streamline your model selection and accelerate your discoveries.
5 Essential Steps for a Successful Immuno-Oncology In Vivo Study
Conducting an Immuno-Oncology in vivo study is a complex process that requires meticulous planning and execution. For oncology researchers and biotech professionals, ensuring the success of these studies is crucial for advancing cancer treatments. Here are the five essential steps to guide you through a successful immuno-oncology in vivo study. 1. Select the Best Tumor Models One of the first critical steps is choosing the appropriate tumor models. This involves: Molecular Data Analysis: Leverage molecular data to select tumor models that express your drug target and most closely represent the target patient population. Ex Vivo Screening: Utilize ex vivo screening techniques to evaluate the effectiveness of your drug in various tumor models and select models with the best response profile. By selecting the most relevant tumor models for your immuno-oncology in vivo study, whether Patient-Derived Xenografts (PDXs) or cell lines, you can ensure that your immuno-oncology in vivo studies will provide more accurate and translatable results. Verified target expression via proteomics and genomics data, and selection of pretreated or naïve models are two extremely important elements of model selection. 2. Choose the Right Mouse Model The choice of mouse model can significantly impact the outcomes of your immuno-oncology in vivo study. Below we provide some directions on how to navigate the different available mouse model options. Murine immune system: Syngeneic Mouse Models: These are generally used when the candidate immuno-oncology drug is able to interact with the murine version of the human target. These models are generated by inoculating murine cell lines into the mice, providing a controlled environment to study immune response. Knock-in Humanized Mouse Models: These models are used when the candidate immuno-oncology drug only interacts with the human target molecule. These models are generated by inserting human genes into the mouse genome, allowing the candidate drug to target the murine immune response. Humanized immune system: Adoptive transfer humanized models: This group of humanized mice allows for the engraftment of mature immune cells such as PBMCs and NK cells. The experimental strategy consists of engrafting human immune cells isolated from peripheral blood in immune-compromised mice to mimic some aspect of the human immune response. CD34 HSC humanized model: This approach provides a platform closer to the human immune system by engrafting CD34 hematopoietic stem cells (HSC) from the human cord blood. The presence of the main populations of the human immune system, including both innate and adaptive immune players, ensures an ideal platform to test a variety of immune-targeting drugs. Selecting the right mouse model for your immuno-oncology in vivo study is crucial for understanding how your treatment will interact with the immune system. (Read our blog "Choosing the RIGHT Model - Syngeneic versus Humanized Mouse Models") 3. Evaluate Drug Toxicity Before proceeding with efficacy studies, it's essential to evaluate the safety of your drug in the chosen mouse model system: Acute Toxicity Testing: Assess the immediate effects of your drug on the mouse models to identify any potential adverse reactions. Usually, a single high dose of the agent is administered to the animals, and toxicity is evaluated within 24 hours from drug administration. Chronic Toxicity Testing: Examine the long-term effects to ensure the drug is safe for prolonged use. In this case, animals are exposed to repeated lower doses of the candidate agent to assess toxicity over a longer period of time. Ensuring the drug's safety through comprehensive toxicity testing is vital for the integrity of your immuno-oncology in vivo study. 4. Evaluate Tumor Killing Next, conduct tumor-killing studies to evaluate the efficacy of your treatment: In Vivo Efficacy Testing: Measure the tumor size reduction and observe the mouse models' overall health. Comparison Studies: Compare your drug’s performance with existing treatments to establish its effectiveness and advantage over standard of care therapies. These studies will provide concrete evidence of your immuno-oncology drug's potential to kill cancer cells, a fundamental step before moving to the clinical research stage. 5. Analyze the Mechanism of Drug Action Understanding how your drug works at a molecular level is the final step to creating a successful immuno-oncology in vivo study. Flow Cytometry: Use flow cytometry to analyze the types and states of cells affected by your treatment. This analysis will reveal the effects of your candidate drug on the number, proliferation, and activation of the different immune cells. (Read our blog "Using Flow Cytometry as an In Vivo Study Endpoint") RNA Sequencing (RNA-seq): Perform RNA-seq to examine gene expression changes induced by your drug. Complementary to flow cytometry, RNA-seq, and DRUG-seq can expose the on-target and off-target effects of your candidate drug, possibly revealing new therapeutic and synergistic opportunities. (Read our blog "Exploring DRUG-seq: Revolutionizing RNA-seq in Oncology Research") These analyses will give you insights into the precise mechanisms through which your immuno-oncology drug operates, paving the way for further development and optimization. By following these five essential steps, you can enhance the accuracy, efficacy, and safety of your immuno-oncology in vivo study. At Champions Oncology, we are committed to supporting researchers with cutting-edge tools and expertise. Ready to take your research to the next level? Reach out to our team of experts and see how we can help you achieve groundbreaking results.
Development of Innovative Therapies Utilizing Preclinical CLL Models
Did you know that Chronic Lymphocytic Leukemia (CLL) is a relentless adversary in the realm of hematologic malignancies? Characterized by the excessive accumulation of abnormal B lymphocytes, CLL presents a unique set of challenges for oncologists, hematologists, and cancer researchers. Despite advancements in targeted therapies and immunotherapies, achieving sustained remission remains elusive for many patients. In this blog, we will explore the critical role preclinical CLL models play in the development of novel therapies. From understanding the disease's biology to facilitating the development of breakthrough treatments, these models are indispensable tools in our fight against CLL. The Role of Preclinical CLL Models in Cancer Research Preclinical CLL models are the unsung heroes of CLL research, providing a crucial bridge between laboratory discoveries and clinical applications. These models enable researchers to study the complex biology of CLL in a controlled environment, offering insights that are impossible to glean from human studies alone. By replicating the disease in animals or in culture with preclinical CLL models, scientists can test the efficacy and safety of potential therapies before they reach clinical trials. This accelerates the drug development process and enhances our understanding of disease mechanisms. In essence, preclinical CLL models are the bedrock upon which modern CLL cancer therapies are built. Overview of Current Preclinical CLL Models The landscape of preclinical CLL models is diverse and continually evolving. Each model offers unique advantages and limitations, making choosing the right one for specific research objectives essential. Cell line-derived Xenograft (CDX) Models Xenograft models involve transplanting human CLL cells into immunodeficient mice. These preclinical CLL models allow for the study of human-specific disease characteristics and the evaluation of human-targeted therapies. However, the lack of a fully functional immune system in these mice limits the study of immune-based treatments. Genetically Engineered Mouse Models (GEMMs) GEMMs are designed to mimic the genetic aberrations found in human CLL. These preclinical CLL models provide a more accurate representation of the disease's progression and response to therapies. They are particularly valuable for studying the genetic and epigenetic factors driving CLL. Patient-Derived Xenografts (PDXs) PDXs involve implanting primary CLL cells from patients into mice and serially passaging to obtain stable in vivo models[1]. These preclinical CLL models retain genetic features of the original tumor, making them predictive of clinical outcomes, although the lack of a proficient immune system needs to be considered. Using Preclinical CLL Models in Oncology Research The latest therapies approved for CLL are a testament to the power of preclinical CLL models in developing revolutionary therapies. Pirtobrutinib (approved by the FDA at the end of 2023[2]) and lisocabtagene maraleucel (approved by the FDA for use in CLL in 2024[3]) were meticulously tested in preclinical CLL models before their clinical debut, ensuring their safety and efficacy in targeting CLL cells. Pirtobrutinib is a highly selective, noncovalent Bruton tyrosine kinase inhibitor (BTKi). Preclinical testing of pirtobrutinib was conducted in several preclinical CLL models. CLL cell lines were used to assess target engagement, potency, cellular phosphorylation, and other cellular activity of the inhibitor[4, 5, 6]. Further studies in primary CLL cells and xenograft models confirmed pirtobrutinib's ability to kill CLL cells and reduce tumor burden[4, 6]. Lisocabtagene maraleucel is a CD19-targeted CAR-T cell therapy. Preclinical studies of lisocabtagene maraleucel involved in vitro and xenograft models to evaluate its ability to target and eliminate CLL cells[7, 8]. These preclinical CLL models provided crucial data on the therapy's potency, specificity, and potential side effects. The success of these preclinical trials paved the way for pirtobrutinib and lisocabtagene maraleucel approval and use in clinical settings. These therapies are now transforming the treatment landscape for CLL and other hematologic malignancies. The Impact and Future of Preclinical CLL Models in Developing Novel Therapies The impact of preclinical CLL models on the development of new therapies cannot be overstated. They have accelerated the discovery of novel treatments, reduced the risk of adverse effects, and improved patient outcomes. However, the field is far from static. Advancements in Model Precision The future of preclinical CLL models lies in their ability to reproduce clinical characteristics and support personalized medicine. The use of primary patient-derived CLL models by direct injection of patients’ cells in mice without additional passages in the animals allows better preservation of tumor heterogeneity and patient population diversity[9], Patient-derived CLL models can be used in a preclinical trial format as well as for the testing of therapies on individual patient tumors, enabling tailored treatment strategies. The development of such models in humanized mice, which possess a reconstituted human immune system, will further improve the robustness of these models as patients’ surrogates by enabling a deeper understanding of how therapies interact with the human immune system, leading to more effective treatments. Integration of Computational Models Integrating computational models with preclinical studies is another promising avenue. By simulating disease progression and treatment responses in silico, researchers can optimize experimental designs and predict outcomes more accurately. This synergy between computational and experimental approaches is poised to accelerate the development of next-generation CLL therapies. The Importance of Continued Research and Innovation Preclinical CLL models are the linchpin of CLL therapy development, providing invaluable insights and accelerating the transition from bench to bedside. As we continue to refine these models and integrate new technologies, the future of CLL treatment looks increasingly promising. Champions Oncology's bank of preclinical CLL models includes primary models derived from pretreated and naive patients. With deep multi-omic and multimodal characterization and comprehensive clinical annotations, we strive to make our preclinical CLL models the best tool to accelerate your drug pipeline through reliable data and extensive expertise in the hematologic malignancies field. Contact us to speak with one of our experts.
Using 3D Ex Vivo Tumor Models for Oncology Research: An Expert Guide
Did you know that 3D organ-like tumor models are biomimetic and yield superior results in drug screening? These models more accurately mimic cell-cell signaling and physiological conditions, providing a superior representation of human tumors outside the body. This blog is dedicated to answering frequently asked questions about Champion's advanced TumorGraft3D platform and assisting scientists in choosing the right platform for their drug screening endeavors. Champions Oncology offers the multiclonal TumorGraft3D drug screening platform, a biologically relevant platform with 3D organoid models derived from our superior, well-characterized patient-derived xenografts (PDXs) for ex vivo drug testing. 1) What is TumorGraft3D and what differentiates them from conventional 3D models? TumorGraft3D models are self-organizing three-dimensional PDX-derived cell clusters that mimic parental human tumor's morphological and molecular phenotype, thereby rendering themselves clinically relevant models for drug discovery. 2) What types of TumorGraft3D models are currently available? Our TumorGraft3D biobank is constantly expanding and currently includes over 150 off-the-shelf models across 16 different tumor types for ex vivo studies. However, our entire PDX bank is available for TumorGraft3D generation, only requiring a short pre-study development phase before becoming available for clients’ studies. 3) What are the advantages of using TumorGraft3D models compared to cell lines or in vivo PDX studies? TumorGraft3D models are generated from our deeply characterized and clinically annotated patient-derived xenograft models and maintain the parent model’s characteristics. They represent the heterogeneity of the patient population and have drug response profiles comparable to the parent PDX model. This makes them a great resource for ex vivo drug screening, with better clinical correlation than cell lines, and have a faster turnaround time and higher throughput than in vivo studies. 4) Are TumorGraft3D models a close representation of patients’ tumors? How do you ensure that patients' characteristics are not lost? TumorGraft3D models are derived from our PDX models without intermediate processing steps and are maintained at low passages to avoid genetic drift and preserve the parent PDX model characteristics. Moreover, IHC, NGS, and drug response analysis are conducted to characterize the TumorGraft3D model and verify that this maintains the parent PDX model’s molecular and pharmacological profile. 5) Why do you use a matrix-free assay? What are the advantages and disadvantages of it? Traditionally, 3D tissue models have been cultured using an extracellular matrix-dependent approach, where the supportive extracellular matrix is derived from natural or synthetic sources. Matrices can pose several challenges such as sourcing of materials, interactions with test agents, and interference in imaging-relevant readouts. Matrix-free approaches allow the end user to observe the self-organization of 3D tissue models without using an exogenous matrix, circumventing all the above challenges. A matrix-free environment allows homogeneous distribution of therapeutic agents and easy access to tumor and immune cells. Additionally, the absence of confounding factors simplifies the assay readout and reduces variability. The use of an exogenous matrix can mimic the physical characteristics of the TME but can also influence tumor cell behavior and it interferes with flow cytometry readouts. 6) How can I use TumorGraft3D to study the TME and agents targeting/engaging the TME? TumorGraft3D models are a well-defined versatile platform that lends itself to various co-culture options and readouts that can generate a complete picture of the TME response to test agents. At Champions, we offer co-culture assays with autologous and allogeneic NK cells, PBMCs, TILs, and other immune cells, allowing for testing of several classes of therapeutic agents such as T cell and NK cell engagers, BiTEs, ADCC drugs, immune checkpoint inhibitors, small molecules, gene therapy, cell therapy, and combinatorial therapy. 7) What are the applications of TumorGraft3D? How can I use this platform to advance my oncology research program? TumorGraft3D models' versatility and clinical relevance, along with their molecular characterization, make them the ideal ex vivo models to measure agent efficacy and on-target effect, unravel the complexities of tumor biology and the crucial interactions within the tumor microenvironment, and evaluate synergistic combination therapies to de-risk in vivo studies through data-driven decisions. 8) How does Champions Oncology ensure the quality of your 3D tumor models? At Champions, we strive to provide the highest quality services. Our TumorGraft3D models are carefully developed following cutting-edge guidelines for organoid development and culture. Our models are: • maintained at low passages to retain molecular and phenotypic characteristics, • qualified for assays using predefined SOC compounds, • verified for true 3D structure formation and viability throughout the length of the assay. 9) What is unique about the TumorGraft3D platform? Specially tailored for integration with various immune cell types, TumorGraft3D models incorporate the unparalleled advantage of Champions’ proprietary autologous systems. Our proprietary autologous co-culture systems proficiently examine the potency of therapeutic agents alongside the intricate interactions between a patient's tumor and their own immune system. This advancement eliminates the inconsistencies commonly seen with the use of allogeneic donors, directing the focus onto patient-specific responses. Our clinically relevant models, coupled with advanced high-content imaging and flow cytometry immunophenotyping make our platform a rich source of insights. With unprecedented precision, scientists can now: • monitor the influence of test agents on the interaction between tumors and their microenvironment, • decrypt their mechanisms of action, • meticulously track the infiltration of immune cells, • and craft potent combination therapies tailored for IO applications.

Navigating Clinical Specialty Testing: Key Insights into Regulatory Compliance
Clinical specialty testing laboratories, like Champions Oncology, are expected to adhere to stringent standards to ensure accuracy and reliability of test results which can have life-altering implications for patients. Regulatory compliance is not a mere bureaucratic hoop but a foundational element that guarantees the integrity of laboratory operations. Navigating through the complex landscape of clinical specialty testing and its regulatory environment is crucial for the success of each clinical trial. Regulatory compliance within each clinical trial is vital to ensure data validity and also ensures each laboratory’s commitment to patients’ safety. In this blog post, we’ll explore the intricacies of adhering to Good Clinical Laboratory Practice (GCLP), Clinical Laboratory Improvement Amendments (CLIA), and College of American Pathologists (CAP) standards, compare regulatory frameworks in the United States (US) versus the European Union (EU) and underscore why meticulous regulatory compliance is a non-negotiable for each clinical trial. Clinical specialty testing laboratories, like Champions Oncology, are expected to adhere to stringent standards to ensure accuracy and reliability of test results which can have life-altering implications for patients. Regulatory compliance is not a mere bureaucratic hoop but a foundational element that guarantees the integrity of laboratory operations. Good Clinical Laboratory Practice (GCLP) GCLP is a quality system that ensures laboratories conducting clinical trial testing provide data of consistent quality. It bridges the gap between the guidelines provided by Good Laboratory Practice (GLP) and Good Clinical Practice (GCP), focusing on pre-analytical, analytical, and post-analytical processes. Clinical Laboratory Improvement Amendments (CLIA) In the United States, CLIA regulations pertain to laboratory testing and require labs to be certified by the federal government. They establish standards for test performance, personnel qualifications, quality control, and proficiency testing for each specific assay performed at the specialty testing laboratory. College of American Pathologists (CAP) The CAP accreditation is an internationally recognized program that provides a framework for clinical labs to achieve excellence in patient care and ensure compliance with statutory and regulatory requirements. CAP takes a peer-reviewed approach to help maintain the highest standard of care. While there may be considerable overlap in what these regulations and standards aim to achieve, there are nuanced differences in their requirements and scopes. GCLP is broader and more flexible in its application, potentially accommodating international guidelines. CLIA is prescriptive and specific to the United States, focusing significantly on the analytical phase of testing. CAP, albeit a US-based program, aligns with many international standards and offers a comprehensive accreditation process that envelopes all aspects of lab operations. Comparatively, the European Union (EU) takes a different approach to laboratory oversight. The EU mandates that each company ensures the quality and safety of its laboratories, but it does not impose a uniform set of standards. Instead of an EU-wide equivalent to CLIA, countries may have their own regulatory frameworks or adhere to international standards like those of the International Organization for Standardization (ISO). Regulatory standards are the pillars that support the validity of clinical trial data. They are key to ensuring that the specialty tests upon which clinical decisions are based are reliable and reproducible. Compliance ensures patient safety, the validity of data submitted to regulatory authorities, and ultimately the success of a clinical trial. Failures in compliance can lead to serious legal consequences and ethical breaches, undermining public trust. Every clinical scientist must understand that regulatory compliance is not simply about following rules; it's about upholding the scientific rigor and ethical duty inherent in clinical research. Each standard, whether it be GCLP, CLIA, or CAP, serves as a QA/QC mechanism to this end. By mastering these regulatory frameworks and recognizing their importance in every aspect of a clinical trial, we safeguard the integrity of clinical research, protect patient welfare, and contribute to the greater good of advancing scientific clinical research.
Accelerating Innovation & Drug Development with Pre-treated PDX Models
The landscape of oncology research and the quest for better treatments have intensified with the advent of precision medicine introducing more targeted agents with higher efficacy and lower toxicity compared to traditional chemotherapy. In personalized medicine, where tailored treatments are revolutionizing patient care, understanding drug resistance mechanisms is key to developing more effective therapies. This blog takes a deep dive into pre-treated patient-derived xenograft (PDX) models, explaining what they are and how they are employed in preclinical studies to combat resistance to standard of care drugs, ultimately leading to better patient outcomes. Refining Precision: PDX Models in a Nutshell PDX models involve the transplantation of human cancer tissue into immunodeficient mice for in vivo growth and expansion. The resultant 'model' is a living tumor system that retains molecular and cellular characteristics closely resembling those of the original patient tumor. Compared to traditional cancer cell line models, PDX models are superior avatars as they capture the heterogeneity and microenvironmental cues found in human tumors. This fidelity allows for a more accurate predictive platform to assess drug responses in preclinical oncology research. Unveiling Resistance: The Power of Pre-treated PDX Models In the quest to outwit cancer, the central challenge has always been predicting and overcoming therapeutic resistance, which often arises after initial treatment with standard of care drugs. Pre-treated PDX models are established from tumors from patients that have previously been exposed to treatments, thus embodying resistant tumor phenotypes that can mimic clinical presentations. The use of PDX models after exposure to specific lines of chemotherapy, hormone therapy, or targeted drug regimens reveals a wealth of insights into how these tumors adapt, evolve, and evade the therapeutic effects. This critical approach allows researchers to identify the genetic and molecular determinants of resistance and devise strategies to combat them. Pre-treated PDX Models at Champions Oncology Champions’ highly characterized biobank includes over 1,000 tumor models across 60+ tumor types derived from advanced stage primary and metastatic patient tumors pre-treated with a plethora of therapies including the latest generations of targeted antibodies and small molecules, immune checkpoint inhibitors, ADCs, bispecific antibodies, and even experimental therapies currently undergoing clinical trial. Our pre-treated models can be easily identified in Lumin, where you can select models of interest to you based on clinical and molecular characteristics, and draft a PDX cohort for your next study. With extensive longitudinal clinical annotations and industry-leading multi-omic and multi-modal characterization, Champions' pre-treated tumor models allow for testing therapeutic agents in systems that closely mirror the clinical trial patient population significantly derisking the drug development process. Transforming Resistance to Resilience in Research Pre-treated PDX models have already catalyzed groundbreaking advancements in oncological research. For instance, studies have utilized these models to identify secondary oncogenic events that drive drug resistance and to develop combinatorial therapies that prevent or even reverse drug insensitivity. One of these studies is highlighted in our case study "Using Champions’ patient-derived xenograft (PDX) models for preclinical validation of HER2-specific small molecule inhibitors" where tucatinib is used as a single agent or in combination with trastuzumab in a selection of HER2-amplified PDX models, both pre-treated and naïve, showing tumor inhibition in trastuzumab-resistant models. The data derived from pre-treated PDX models can be integral in informing personalized treatment strategies for cancer patients. The Future of Research and Care in Oncology The implications of utilizing pre-treated PDX models are not just confined to the benches of research labs. These models have the potential to significantly impact the course of clinical trials, where they can serve as powerful tools to validate therapeutic responses and study mechanisms of drug failure in a more controlled environment. Investigating drug resistance in PDX models allows for a more systematic and accelerated approach to drug development. New compounds can be tested on pre-treated PDX models to evaluate their potential benefits, potential interactions with existing therapies, and their ability to overcome resistance, ensuring a fast track to clinical trial. By providing a more accurate representation of human tumors and treatment responses, pre-treated PDX models bridge the gap between bench-side discoveries and clinical applications. This synergy is crucial in ensuring that the most promising treatments reach patients in a timely and effective manner.

Deciphering CNV: Utilizing Gene Copy Number Variation Data in Lumin
Gene copy number in a tumor cell is a significant indicator of the implication of a given gene in several oncogenic processes such as uncontrolled proliferation, elusion of programmed cell death, and resistance to treatments. At Champions Oncology, gene copy number analysis is performed by using the EXCAVATOR2[1] tool on whole exome sequencing (WES) data generated to characterize our patient-derived xenograft (PDX) models. This tool allows for classifying each segmented region into five qualitative genomic states (two-copy deletion, one-copy deletion, normal, one-copy duplication, and multiple-copy amplification) and quantifying the number of chromosomal copies. All our model characterization data can be explored in Lumin, a unique solution integrating Champions’ tumor model multi-omic data and public datasets in one accessible platform for model selection and data interpretation. In Lumin, gene copy number analysis results are presented in the format shown in the example below: Here we answer the most common questions about CNV data reporting to help you navigate WES data in our Lumin platform as well as interpret your own study data. Q: What does Log2R mean and why sometimes is it marked as NA? A: The Log2R (Log2 ratio) value represents the number of copies relative to the normal reference sample (NA12878). The EXCAVATOR2 algorithm used to calculate CNV uses a median normalization approach, with the log-transformed ratio (Log2R) being calculated from the window mean read count (WMRC) values of the test sample compared to the normal reference. When Log2R value is marked as NA, no significant copy number alteration was detected. Q: What are the Call values and how are they defined? A: The Call value is calculated using the FastCall algorithm[2] and classifies each segmented region as one of five possible states: 2 copy deletion= -2; one copy deletion= -1; normal= 0; one copy duplication= 1; and multiple copy amplification= 2. Q: Are the copy number values the absolute or the relative copy numbers detected? A: Copy number values represent the absolute copy number detected, which is derived from the Copy Number Fraction and is rounded to the nearest integer. Q: Do “Amp” and "Del" always mean that there is a gain or loss in copy number? Or is this only the case if the “Alteration” column says “Gain” or “HomoDel”/ “Hetloss”? A: The "amp_del" column definitions are derived from the Call values, whereas the "Alteration" column classifies the alteration based on the copy number detected. In some instances, there may be discordance between the copy number and Call (as shown for FAM231C gene in the example table above), as the two values are derived by approximation of continuous values. In this specific example, the conflict between the two annotations is indicative of the tumor gene copy number being between 1 and 2, suggesting the presence of both cells with 1 and cells with 2 copies of that genomic region. For additional investigation, we recommend looking at the continuous values in the raw data. Q: What columns should I consider when I want to search for a model with an amplification/deletion for a certain gene? A: We would first recommend using the "CopyNumber" column to identify models with an amplification or deletion of a specific gene. Once you have filtered the models based on this, you can then use the “Alteration” column to verify whether Call, copy number, and the “amp_del” column values are concordant.
The Evolution of Treatment in TNBC: A Closer Look at Pembrolizumab
In the realm of oncology, few words instill as much uncertainty and trepidation as "triple negative breast cancer" (TNBC). With its resistance to many standard forms of therapy, TNBC demands a new generation of treatment innovation. Enter pembrolizumab—an immunotherapy designed to engage the body’s immune system in the fight against cancer. Its recent breakthroughs in clinical trials have charted a promising new course in the treatment of this aggressive form of breast cancer. The TNBC challenge Conventionally, breast cancer treatment plans are designed around the presence or absence of three receptors: estrogen, progesterone, and HER2/neu. TNBC, characterized by the absence of these receptors, compels a more bespoke approach, given the limitations it imposes on targeted treatments available for other forms of breast cancer. The lack of defined therapeutic targets has historically left TNBC patients with fewer options and a greater risk of disease progression and poor prognosis.[1] The arrival of pembrolizumab: a precision tool in the TNBC arsenal Pembrolizumab operates on a fundamentally different principle than traditional treatments. It is an immune checkpoint inhibitor that effectively releases the brakes on the immune system, allowing it to identify and combat cancer cells in a manner that is highly specific to a patient's tumor profile. This tailored approach has been nothing short of revolutionary in cancers with high mutational loads, such as TNBC, where the potential for an immune system response is significant.[2] Groundbreaking trials: pembrolizumab's journey to TNBC approval KEYNOTE-355 The KEYNOTE-355 study burst into the scientific limelight with its findings on the efficacy and safety of pembrolizumab in combination with chemotherapy in the treatment of locally recurrent inoperable or metastatic TNBC. The trial's incorporation of a diverse patient population, coupled with the comprehensive analysis of the drug's performance set a new precedent for the depth and breadth of oncology research.[3] This landmark study, along with other trials such as KEYNOTE-012 and KEYNOTE-086, has served as evidence in the FDA's approval of pembrolizumab in combination with chemotherapy for the treatment of advanced TNBC expressing high levels of PD-L1. These studies have demonstrated not only the drug's ability to improve patient outcomes but also its potential to revolutionize the treatment landscape for this challenging form of breast cancer.[4] The KEYNOTE-355 study has been a beacon of hope for those combating triple-negative breast cancer, illuminating the path forward with its groundbreaking results. Central to its findings is the remarkable improvement in overall survival (OS) rates for patients treated with pembrolizumab in conjunction with chemotherapy. Specifically, the study delineates a median OS of 23 months for these patients, significantly surpassing the 16.1 months median observed in those receiving chemotherapy alone. This stark contrast not only emphasizes pembrolizumab's efficacy but also marks a tangible advancement in extending the lives of individuals facing this formidable adversary.[4,5] KEYNOTE-522 Similarly, the KEYNOTE-522 trial, which investigated pembrolizumab in the neoadjuvant setting, demonstrated a significant pathologic complete response (pCR) rate. This paradigm-shifting evidence supported the FDA's approval of pembrolizumab for high-risk early-stage TNBC, leading to a pivotal shift in the narrative around treating this formidable adversary.[6] The results from KEYNOTE-522 have been nothing short of revolutionary, illustrating a marked improvement in both pCR (7.5% higher than in the control arm) and event-free survival (EFS). Patients on the pembrolizumab arm experienced EFS benefit regardless of tumor PD-L1 status.[6,7] What makes these results particularly compelling is the implication for long-term survival outcomes. Early indications suggest that the increase in pathologic complete response rates correlates with longer overall survival, offering new hope that pembrolizumab could extend lives in a population historically challenged by high relapse and mortality rates. These outcomes underscore the importance of pembrolizumab in the TNBC treatment paradigm, highlighting its potential to significantly alter the prognostic outlook for patients facing this aggressive form of breast cancer.[6,7] The future of the TNBC treatment landscape The integration of pembrolizumab into TNBC treatment protocols is a landmark event that underscores the ongoing refinement of personalized oncology care. While the successes in clinical trials are incredibly encouraging, the implementation of these new standards into broader clinical practice requires deliberate consideration of patient selection, administration protocols, and the management of potential immune-related adverse events.[8] Researchers and clinicians are now tasked with harnessing the full potential of this breakthrough treatment as well as identifying novel strategies to extend patient survival and improve quality of life. The future of TNBC treatment lies in leveraging emerging technologies and collaborative frameworks to uncover even more effective therapeutic solutions.
Immune Checkpoint Blockade Strategies in Renal Cell Carcinoma
Renal cell carcinoma (RCC) is a common cancer of the genitourinary tract that has very poor survival outcomes if metastatic. RCC is now understood to be composed of several different types of cancer with different genetic features and varied clinical responses. Histological diagnosis has been the primary method to diagnose RCC and has been used to define three major RCC subtypes, including the most common subtype, clear cell renal cell carcinoma (ccRCC), papillary renal cell carcinoma (PRCC; further divided into two subtypes), and chromophobe renal cell carcinoma (ChRCC)[1]. More recent comparative genomic and phenotypic analysis has identified mutations and epigenetic modifications associated with different histological subtypes[2]. Across all subtypes, increased DNA hypermethylation and gene alterations in CDKN2A were associated with a poor prognosis as was an increased Th2 immune gene signature. For ccRCC, increased levels of mRNA transcripts associated with ribose metabolism and the immune response were associated with poor survival. ccRCC is also defined by the early loss of chromosome 3p, which in turn causes a loss of heterozygosity for the VHL, PBRM1, SETD2, and BAP1 tumor suppressor genes and subsequent mutation of these genes that leads to tumorigenesis[3]. There is also a subset of ChRCC with a unique metabolic expression pattern that is associated with extremely poor survival[2]. PRCC can be classified as type 1, for which PBRM1 mutations are linked to poor survival but type 2 PRCC has increased expression of glycolysis and metabolism-related mRNA transcripts[2]. ccRCC tumors with VHL mutations show overexpression of vascular endothelial growth factor (VEGF) and hypoxia-inducible factors (HIFs) can contribute to angiogenesis and cancer progression. Similarly, some RCCs show hyperactivation of the serine/threonine kinase mammalian target of rapamycin (mTOR), which can lead to the overproduction of VEGF. VEGFR inhibitors and anti-VEGF antibodies have been tested as therapies for RCC, and the anti-VEGF antibody bevacizumab has been approved for use in combination with IFN-α for metastatic RCC[4]. The mTOR inhibitors everolimus and temsirolimus have also been approved for the treatment of RCC, typically in combination with tyrosine kinase inhibitors (TKIs)[5]. Combination therapies that target VEGF and mTOR are considered more effective since they work in concert to target tumor growth and vascularization, whereas sequential treatments are typically associated with a greater likelihood of tumors developing resistance[6]. Unfortunately, these combination therapies are associated with undesirable toxicities and have not been linked with durable responses[7]. Notably, an HIF-2α inhibitor, belzutifan, has shown good overall response rate and duration of response and has been approved by the FDA in 2021 for use in adults with VHL-associated RCC and in patients with advanced RCC who have been treated with anti-VEGF therapy and anti-PD-1/PD-L1 therapy[8]. Combination therapies with belzutifan and other targeted agents or ICB are currently being evaluated in clinical trials[9]. Advances in immunotherapy have led to transformative treatment options for RCC. Immune checkpoint blockade (ICB) has been one promising treatment strategy, and the FDA approved the use of anti-PD-1/PD-L1 (nivolumab) for the treatment of advanced ccRCC in 2015[10]. A follow-up study showed that anti-PD-1 was associated with the best clinical benefit in ccRCC carrying loss-of-function mutations in PBRM1, which appears to affect tumor expression profiles in such a way to maintain responsiveness to checkpoint blockade[11]. Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) is another checkpoint molecule targeted for checkpoint blockade with the monoclonal antibody ipilimumab. Similar to PD-L1 blockade, CTLA-4 blockade was associated with a partial response against metastatic RCC. Combination therapies have shown much more durable responses in patients with advanced RCC, and the FDA approved the use of nivolumab plus ipilimumab for the treatment of intermediate or high-risk metastatic RCC in 2018[12]. ICB in combination with TKI have also been approved for advanced disease [13,14,15,16]. Unfortunately, PD-1/PD-L1 blockade has been less successful for PRCC[17] and ChRCC[18], and further studies are needed to identify therapeutic targets in these forms of RCC. Clinical studies with other checkpoint blockade targets are currently underway and are likely to provide new treatment options to RCC patients[19]. The future of RCC therapies relies on the identification of new molecules or pathways in tumor cells that can be targeted therapeutically without causing toxicity or promoting resistance. Advances in single-cell omics are leading the way in terms of target identification and understanding how different forms of RCC progress.
Choosing the RIGHT Model - Syngeneic versus Humanized Mouse Models
Mouse models have been the workhorses of preclinical immuno-oncology (IO) research, and advances in mouse model development have expanded to applications for nearly all types of solid tumors and hematological malignancies. Preclinical evaluation of experimental immunotherapies has been advanced by syngeneic and humanized mouse models. Syngeneic mice are one of the most established types of models used in cancer research, whereas humanized mice are a contemporary mouse model that has been critical to the screening of immunotherapeutic agents. Here we highlight features of syngeneic and humanized mouse models and define which models are most relevant to different phases of preclinical IO research. Syngeneic Tumor Models Syngeneic tumor models are created by transplantation of tumor cell lines into immunocompetent mice with the same genetic background as the cell line.[1] Tumors can be transplanted intravenously or subcutaneously into mice and typically grow rapidly over several weeks. Different types of tumor cell lines can be used in this type of model, including spontaneous, transgenic, or carcinogen-induced tumor cell lines. Syngeneic mouse models are best suited for screening novel IO agents or gaining insight into anti-tumor responses in the context of an intact immune system. Given the rapid growth of tumors in syngeneic mice, these models are less well suited to studying early events in tumor growth associated with cancer stem cells or understanding the contributions of heterogeneous tumor microenvironments, and these models typically do not recapitulate the mutational heterogeneity observed in human tumors.[2] Champions Oncology offers an extensive suite of syngeneic models, meticulously curated to support your groundbreaking investigations within a robust and intact immune environment. We harness advanced biomedical techniques to deliver comprehensive data with accurate and reliable experimental endpoints: cutting-edge flow cytometry for detailed characterization of immune cell populations, western blot analysis to quantify the expression of pivotal cancer and immune signaling proteins, immunohistochemistry for the precise localization of these proteins within the tissue architecture. Explore our syngeneic tumor model offering here. Humanized Tumor Models Humanized tumor models are a more recent addition to preclinical IO research that provide valuable insight into how individual tumors from patients (xenografts) respond to experimental therapies. Prior to the development of humanized mouse models, human xenograft models were used for screening cytotoxic or immunotherapeutic agents like chimeric antigen receptor (CAR) T cells[3], and these models use human tumor cell lines or patient-derived specimens transplanted into immunocompromised host mice. Different immunocompromised models can be used, including athymic mice that lack T cells or severe combined immunodeficiency (SCID) models that lack all adaptive immune responses. Humanized mice have been engineered from immunocompromised mouse strains that include genetic mutations in other adaptive immune functions that allow for the engraftment of human hematopoietic cells. The NOD/SCID IL2rγ chain knockout (NSG) mouse (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) is one of the most used combined immunodeficiency models that can be engrafted with human hematopoietic cells and primary human tumors.[4] These patient-derived xenograft (PDX) models are useful for evaluating experimental IO therapies in the context of the human immune system and can use human immune cells from the same or different donor as the tumor source. PDX models are suited to evaluating experimental therapies in the context of a genetically heterogeneous tumor and better recapitulate aspects of the tumor microenvironment. (Download our case study "Using Champions' Prostate PDX Models for Preclinical Validation of a Novel, PSMA-directed BiTE" to see how Amgen used our humanized models to test a novel PSMA-targeted BiTE) Tumors can be grafted either orthotopically or subcutaneously and this also impacts how tumors grow and respond to experimental treatments.[5] Given the heavily modified nature of the NSG immune system, these models do not always reflect responses observed in humans during clinical trials. Nonetheless, NSG mice and similarly modified humanized mice offer valuable insights into the efficacy of IO candidates. Champions Oncology's state-of-the-art humanized mouse models are tailored to harness the power of the human immune system. Our portfolio offers a suite of options crafted to empower your scientific investigation: engraftment of cell therapies, adoptive transfer of human PBMCs and NK cells, engraftment of CD34+ hematopoietic stem cells from cord blood for full reconstitution of the human immune system. Learn more about our humanized models offer here. Mouse models are constantly being refined and improved to better reflect human physiology. Both syngeneic and humanized mouse models serve as valuable tools for preclinical IO research and accelerate the screening and evaluation of novel therapeutics.
Advancing the Battle Against Mantle Cell Lymphoma
Mantle cell lymphoma (MCL) is a rare and aggressive non-Hodgkin’s lymphoma (NHL) that originates in B cells and occurs in secondary follicles of the lymph nodes.1 MCL accounts for roughly 6% of all lymphomas diagnosed annually, and despite its relatively low incidence, the aggressive course of MCL means it can quickly become a life-threatening disease. Understanding the origin and progression of MCL is crucial to identify new targets for drug development studies. Understanding Mantle Cell Lymphoma Pathology Over the last decade, our understanding of MCL pathogenesis has evolved from a single definition to a consensus that MCL development and progression is related to a range of molecular events. Early on MCL was defined as a pathognomonic chromosomal translocation t(11;14)(q13;q32) causing a mutation in the CCND1 gene, resulting in the over-expression of Cyclin D1.1, 2 Under normal conditions, Cyclin D1 is heavily regulated and modulates cell cycle transition from G1 phase to S phase.1 However, overexpression of Cyclin D1 activates cyclin-dependent kinases (CDK) 4 and 6 which later deactivate the retinoblastoma protein (Rb), a cell cycle inhibitor. This series of events accelerates cell cycle progression from G1 to S phase in many cell types, including B-cells.3 The progression of this process induces uncontrolled B-cell proliferation causing an enlargement of lymph nodes and immune system dysfunction that eventually spreads to critical organs. Fewer Mutations Raise More Questions Interestingly, Cyclin D1 mutations are not found in all MCL cells, suggesting other molecular actors, such as transcription factors, are involved. In 90% of MCL patients, transcription factor SOX11 is over-expressed in MCL cells with and without mutated CCND1. While SOX11’s role in MCL is not well understood, studies suggest that PAX-5, a transcription factor that regulates B-cell development and differentiation, is activated by SOX11.4 The overexpression of SOX11 that is commonly seen in MCL patients can therefore lead to reduced B-cell differentiation and increased B-cell antigen signaling.3, 5 Identifying Treatment Targets in Mantle Cell Lymphoma B-cell proliferation and differentiation rely on B-cell receptor (BCR) signaling and are activated in response to antigen binding.6 Using the framework for CLL pathogenesis, preliminary MCL-BCR research has found that BCR signaling is highly active in MCL patients and intentional BCR activation resulted in an increase of BCR signaling in MCL cells.7 Additionally, murine studies have found SOX11-overexpressing B-cells have high levels of BTK, a key enzyme involved with BCR signaling, resulting in proliferation which suggests SOX11’s deeper role in MCL.7 Initial MCL treatment includes R-CHOP therapy, however, many MCL patients are refractory or become resistant creating a need for additional therapies. As with CLL, BCR signaling plays a critical role in MCL progression shifting treatment protocols towards BTK inhibitors (BTKi) like ibrutinib, approved in 2013.1, 3 Ibrutinib is metabolized in the liver via CYP3A and CYPRD6 and irreversibly binds to cysteine residue 481 found on the active site of BTK. While this targeting strategy effectively blocks BTK signaling limiting MCL cell development, ibrutinib has significant off-target consequences, namely IL-2 inducible kinase (ITK), which sparked the development of zanubrutinib.8 Zanubrutinib was approved for use in MCL patients and though it has a similar mechanism of action to ibrutinib, its reduced affinity for ITK, a major T-cell and natural killer (NK) cell regulator,9 makes it a highly selective and potent BTKi with improved overall response rate relative to ibrutinib.8 Resistant Mantle Cell Lymphoma and Future Therapeutic Strategies Unfortunately, 32% of MCL patients become resistant to BTKi due to BTK mutations.10, 11 As a result, combination therapies such as ibrutinib combined with cirmtuzumab (a novel anti-ROR1 monoclonal antibody) or with obinutuzumab and venetoclax have been used to treat resistant MCL patients. 11 These therapeutic combinations have shown excellent remission results paving the way for other combination therapies, such as acalabrutinib, rituximab, and bendamustine. 11, 12 Notably, a phase 1 study investigating acalabrutinib, rituximab, and bendamustine achieved an 85% overall response rate which has led to a larger, ongoing Phase 2 study (NCT04115631). Additionally, novel MCL-1 inhibitors are under development in preclinical studies.13 Recently, the FDA has approved the first CAR T cell therapy for relapsed/refractory MCL, brexucabtagene autoleucel (Tecartus).14 Tecartus has shown durable response at the 3-year follow-up even in high-risk patients (NCT02601313), with sustained survival and a 67% complete response rate.15 Understanding the pathophysiology of naïve and BTKi resistant MCL is crucial to developing curative MCL therapies. While standard of care treatments like ibrutinib and zanubrutinib have had some success, additional studies investigating strategic combination therapies are underway providing hope for patients battling this disease. Champions supports your in vivo preclinical studies with low passaged MCL PDX models available for subcutaneous modeling and fully characterized with NGS data in 4 models CTG-3771, CTG-3772, CTG-3776 (shown to be rituximab and ibrutinib resistant) and CTG-3808. Preclinical hematological scientists need to evaluate their pipeline of therapeutic candidates in robust hematological screening platforms; Champions' VitroScreen platform can advance potential next-generation therapies into the clinic, generating additional options for MCL patients.
Exploring DRUG-seq: Revolutionizing RNA-seq in Oncology Research
Oncology research is a field that continuously demands cutting-edge technologies to unravel the complexities of cancer. Among these innovations, RNA sequencing (RNA-seq) has emerged as a vital tool, enabling us to study the transcriptome with unprecedented depth. However, a new star is on the rise in the realm of RNA-seq — DRUG-seq. This application dazzles oncology researchers and genomics professionals with its cost-effective approach, reduced bias, and high sample efficiency, bringing with it a promise to redefine how we investigate cancer at a molecular level.[1] Champ-seq is Champions's DRUG-seq platform. In this comprehensive exploration, we'll dissect the advantages of DRUG-seq, and map out its pivotal role in oncology research, shedding light on its unique applications and use cases in the fight against cancer. Unveiling DRUG-seq: The New Era in RNA-Seq RNA-seq, widely employed for quantifying gene expression, has been a game-changer in understanding the genetic mechanisms driving cancer. However, DRUG-seq takes this a step further, providing a powerful lens through which to examine the transcriptome under a spectrum of conditions, particularly those relevant to drug treatment responses. While traditional RNA-seq methods offer insights into the steady state of gene expression, DRUG-seq provides a dynamic view that captures the immediate and prolonged gene expression changes following drug administration. This level of detail is especially critical in oncology, where the intricate interplay of genetics, environmental factors, and treatment response paints a highly complex picture of disease progression and therapy outcomes. By enabling high-throughput profiling of the transcriptional response to drug compounds, DRUG-seq stands out as a catalyst for precision medicine, biomarker discovery, and the personalization of cancer treatment strategies.[1] The Advantages of DRUG-seq in Comparative Analysis Cost-Effectiveness One of DRUG-seq's most touted benefits is its cost-effectiveness. The methodology uses a smaller number of sequences to cover the transcriptome, thanks to its selective enrichment of pre-existing reference indices. This focus on specific gene regions, associated with drug response or otherwise, lowers the overall sequencing cost per sample, making large-scale comparative studies feasible within more restrained budgets. Sample Efficiency With oncology often facing constraints of sample availability, the high efficiency of DRUG-seq is a game-changer. It requires smaller amounts of starting material, which not only conserves precious samples but also aligns with the trend toward microsampling in emerging clinical research practices. DRUG-seq in Action: Enhancing Oncology Research and Development Drug Response Profiling An immediate application of DRUG-seq is in profiling the response of cancer cells to different compounds. By comparing the expression profiles pre- and post-treatment, researchers gain a comprehensive view of how drugs affect gene networks. This deep understanding underpins the development of more effective therapies by identifying compounds that selectively target critical pathways in specific cancer types.[2] Identifying Novel Drug Targets DRUG-seq empowers the hunt for new targets by revealing unsuspected links between gene expression patterns and drug effects. This insight into the cellular response can lead to the discovery of novel molecular targets that modulate sensitivity or resistance to treatment, providing fertile ground for the next generation of anti-cancer compounds. Biomarker Discovery Precision oncology heavily relies on the discovery and validation of biomarkers to predict patient outcomes and guide therapeutic decisions. DRUG-seq, with its ability to uncover gene signatures indicative of treatment response, plays a pivotal role in biomarker discovery, potentially leading to tests that can stratify patients for tailored treatment interventions.[1] Tumor Heterogeneity Cancer is not a single disease but a collection of disorders, each with its own molecular profile and behavior. DRUG-seq's power to unravel drug response within this context is invaluable, as it allows researchers to study how different cell populations within a tumor respond to treatment. This understanding of tumor heterogeneity can inform the development of combination therapies that target multiple facets of the disease. Conclusion: The Bright Future of high-throughput transcriptional profiling with Champ-seq in Oncology Research From cost-effectiveness and reduced bias to sample efficiency and rich data, Champ-seq provides a valuable addition to the oncologist's toolkit. Its applications span across drug and biomarker discovery, as well as understanding tumor heterogeneity, which is pivotal in the era of personalized medicine. As we have navigated the many advantages, it's clear that Champ-seq isn't just a new trend; it's a technological leap that can redefine the standard for RNA-seq applications in oncology research. By harnessing this tool, researchers can explore cancer treatments with unprecedented precision and efficacy, ultimately leading to improved patient outcomes and a more comprehensive arsenal against this formidable foe.

Developing Flow Cytometry Assays in Non-Human Primates (NHPs)
Non-human primates (NHPs) continue to be a valuable resource for preclinical research because of the similarities they share with humans. NHPs, especially rhesus macaques, are used in preclinical studies for evaluating new drugs or vaccines for safety and efficacy. Flow cytometry assays can be easily adapted to study cells from NHP. Consider these three factors if you are planning to adapt a flow cytometry assay for use with NHP samples. Staining panel prep: Like human subjects, PBMCs can be collected from Non-human primates and evaluated by flow cytometry. Many immune cell subsets, especially B and T cells, share similar phenotypes with their human counterparts. Human antibodies are most often used to stain NHP cells because of similarities shared between surface markers. Consider which cells of interest you may want to study and how existing antibodies used in flow cytometry may be used in your panel. Real-world variability: Many scientists are accustomed to working with an inbred mouse strain, which reduces background variability observed in experimental settings. In contrast, NHPs are outbred animals, and like humans, may display widely varying responses. As you develop your NHP protocols, consider how you will handle highly variable data, including flow cytometry data. These considerations will inform how many animals may be used in each group and how many cells are stained and evaluated in each sample. Metabolism and toxicity: NHPs share many physiological similarities to humans and are a valuable model for pharmacokinetic and pharmacodynamic measurements of experimental drugs and biologics. Toxicity testing can also be done in NHPs and together these data are critical to evaluating candidates in preclinical pipelines. Developing NHP protocols from inception to sample analysis requires working with experts who have experience writing protocols that are compliant with appropriate oversight committees, such as institutional animal care and use committee offices. Working with experts, such as contract research organizations, also assures that any NHP flow cytometry studies will satisfy any necessary regulatory compliance determinations related to drug and biological development.
Triplet Therapy for IDH1-Mutant AML Tumors
Acute myeloid leukemia (AML) is the most common leukemia among adults and has been challenging to treat with modern therapies.1, 2 This disease is highly heterogeneous and characterized by the rapid proliferation of undifferentiated myeloid cells that accumulate within the bone marrow. Several mutations and epigenetic abnormalities characteristic of AML have been targeted through chemotherapies or molecular inhibitors. Fortunately, the relentless pursuit of innovative, effective AML therapies has led to a deeper understanding of AML pathogenesis, such as the role of isocitrate dehydrogenase (IDH) mutations.3 IDH mutations have unique properties that, if properly targeted, may reshape AML patient outcomes and survival rates. Unstable Gene Expression The IDH family consists of three isoenzymes (IDH1, IDH2, and IDH3) and has an important role in the biosynthesis of metabolites involved in the tricarboxylic acid (TCA) cycle. Notably, IDH1 functions as a catalyst for reversible conversion of isocitrate to α-ketoglutarate in the cytosol as well as peroxisomes, yielding 1 NADPH. Downstream, α-ketoglutarate is reduced to D-2-hydroxyglutarate (D-2-HG) as part of the TCA cycle.3 Mutations in IDH1 are present in ~20% of AML patients and result from a single amino acid substitution at Arg132.4 This mutation induces an end-product shift that reduces α-ketoglutarate to R-2-HG instead. Unlike D-2-HG, R-2-HG competitively inhibits α-ketoglutarate-dependent enzymes, such as the TET family, and upon accumulation leads to impaired cellular differentiation and deregulation of DNA methylation. 3, 5 This alteration of DNA methylation and ultimately gene expression activates oncogenes and deactivates tumor-suppressor genes.6 The destabilizing features of IDH1 mutants have thus made this isoenzyme an interesting target for AML therapies. IDH Focused Therapies Since the identification of IDH mutations in 2008, the Food and Drug Administration (FDA) has approved two therapeutics for AML patients with IDH mutations, ivosidenib and enasidenib. Ivosidenib is a reversible, selective inhibitor that binds to the active site of mutated IDH1 to prevent the production of R-2-HG.7, 8, 9 Reduction in R-2-HG reportedly increases D-2-HG concentrations by 100x and restores DNA methylation and cellular differentiation in AML patients.10, 11 Though successful, resistance to ivosidenib and IDH inhibition has emerged, underlining the need for combination therapies that prevent IDH resistance.12 Triplet Therapy There are no triplet regimens currently approved for use in AML, however, clinical trial results have demonstrated successful remission in relapsed and naïve AML patients.13, 14 For instance, an ongoing phase Ib/II study examining ivosidenib and venetoclax with or without azacytidine has shown successful remission in patients with AML IDH1 mutations.13 The durable response to this therapy is especially promising, as the safety profile of this triplet therapy is similar to doublet therapies such as azacytidine + venetoclax and azacytidine + ivosidenib.13, 15 The median event-free-survival (EFS) for patients treated with this new triplet therapy was 36 months13, while previous azacytidine + ivosidenib combination treatments have achieved median EFS or 24 months.16 The Therapeutic Pipeline Prolonged EFS in AML patients receiving ivosidenib in combination with AML standards of care, such as venetoclax and azacytidine, has recently gained the attention of the European Commission (EC). In 2023, the EC announced the approval of ivosidenib in combination with azacitidine for AML patients with an IDH1 R132 mutation.18 Further, clinical studies evaluating the side effects and appropriate dosages of ivosidenib and venetoclax with or without azacytidine are underway. As more clinical data is released, access to more robust therapeutic treatments will become available to improve AML patient outcomes.
Providing Optimal Clinical Care for Cholangiocarcinoma Subtypes
Cholangiocarcinoma is a type of cancer that originates from cholangiocytes, the cells that constitute the bile ducts in the liver, which carry bile from the liver to the small intestine. It is a rare and aggressive cancer that can be categorized into different subtypes based on histological and molecular characteristics. Histologically, cholangiocarcinoma can be classified as intrahepatic, perihilar, or distal. Intrahepatic cholangiocarcinoma originates within the liver, while perihilar and distal cholangiocarcinoma develop in the ducts outside the liver. These subtypes have distinct clinical presentations and treatment approaches [1,2]. In this blog, discover the diverse nature of cholangiocarcinoma subtypes and learn about the specific clinical care required for each subtype. Histological Characteristics and Clinical Care Histological characteristics play a significant role in guiding clinical care for cholangiocarcinoma subtypes. Intrahepatic cholangiocarcinoma often presents as a solitary mass within the liver, while perihilar cholangiocarcinoma typically involves the bifurcation of the bile ducts. Distal cholangiocarcinoma commonly manifests as a tumor in the lower part of the bile duct near the small intestine. The histological features of each subtype influence the choice of diagnostic tests, surgical interventions, and other treatment modalities. For example, surgical resection is often the primary treatment option for intrahepatic cholangiocarcinoma, while perihilar cholangiocarcinoma may require a combination of surgery and liver transplantation. Distal cholangiocarcinoma may be treated with surgery, radiation therapy, or systemic chemotherapy. By understanding the histological characteristics of cholangiocarcinoma subtypes, healthcare professionals can provide appropriate clinical care to improve patient outcomes. Cholangiocarcinoma Subtype-Specific Therapies Each cholangiocarcinoma subtype requires specific therapies tailored to its unique characteristics. For intrahepatic cholangiocarcinoma, surgical resection is often the primary treatment approach, followed by adjuvant therapy such as chemotherapy or radiation therapy. Liver transplantation may be considered in selected cases. Perihilar cholangiocarcinoma, on the other hand, often requires more complex surgical interventions, such as liver resection combined with bile duct resection and reconstruction. Liver transplantation may be an option for patients with advanced disease or underlying liver disease. Distal cholangiocarcinoma may be treated with surgical resection, radiation therapy, or systemic chemotherapy. The choice of treatment depends on the extent of the tumor and the patient's overall health. By tailoring therapies to the specific cholangiocarcinoma subtype, healthcare professionals can optimize treatment outcomes and improve patient survival rates. Molecular Profiling and Treatment Strategies In addition to histological characteristics, molecular profiling has emerged as an essential tool for understanding cholangiocarcinoma subtypes and guiding treatment strategies. Molecular profiling involves analyzing the genetic and molecular alterations in cancer cells to identify potential targets for therapy. Advancements in molecular profiling techniques have allowed researchers to identify specific biomarkers and genetic mutations associated with cholangiocarcinoma subtypes. This information helps in developing targeted therapies that can effectively inhibit the growth and spread of cancer cells. Two targeted therapies are currently available for patients presenting tumors with FGFR2 fusions/rearrangements (infigratinib) or IDH1 (ivosidenib) mutation [3,4]. In general, treatment strategies for cholangiocarcinoma subtypes may include targeted therapies, immunotherapy, chemotherapy, and radiation therapy. Molecular profiling enables healthcare professionals to select the most appropriate treatment options based on the molecular characteristics of the tumor and the patient's overall health [5]. Future Directions in Cholangiocarcinoma Care The field of cholangiocarcinoma care is rapidly evolving, with ongoing research and advancements in treatment options. Efforts are being made to improve early detection methods for cholangiocarcinoma. Early diagnosis allows for timely intervention and increases the chances of successful treatment. Additionally, advancements in precision medicine and molecular profiling techniques hold promise for tailoring treatment plans based on the unique molecular characteristics of each patient's tumor. In conclusion, the diverse nature of cholangiocarcinoma subtypes necessitates specific clinical care for optimal treatment outcomes. Through a comprehensive understanding of histological and molecular characteristics, healthcare professionals can provide personalized therapies and contribute to ongoing research efforts aimed at improving cholangiocarcinoma care.
A Needle in a Haystack: Finding Rare AML Populations by Flow Cytometry
Hematologic malignancies include a wide array of lymphomas and leukemias that affect different immune cell subsets. Acute myelogenous leukemia (AML) is one of the most commonly occurring leukemias in adults and children. AML is a highly heterogeneous disease that can be caused by spontaneous gene mutations or chromosomal translocations, which result in the proliferation of dysfunctional myeloid cells. Cytogenetic and morphologic analyses have been the gold standard methods used in AML diagnosis. Still, flow cytometry-based protocols are becoming more widely used and validated as complementary diagnostic methods that can be coupled with these analyses to better guide treatment plans. Flow cytometry has also become an essential tool to understand AML progression and develop and evaluate novel therapeutics. Consider these aspects of flow cytometry-based analysis of AML for exploratory or preclinical research: Phenotype matters: Immunologists know a lot about what a normally developing myeloid cell should look like in terms of its immunophenotype. Changes in expression of different lineage markers correlate strongly with AML progression and are characterized by the presence of blasts (leukemic cells) in the bone marrow. Flow cytometry-based immunophenotyping offers researchers a rapid and sensitive method for detecting blasts at the onset of disease as well as monitoring changes in this population throughout an experimental therapy. Rare cells from precious samples: A major consideration of following AML progression is the ability to detect relatively rare cells in bone marrow aspirates. This type of sample may be relatively small and is typically used fresh for morphologic evaluation, but even a small volume of remaining aspirate can be used for flow cytometry-based methods that are sensitive and robust enough to detect blasts. Paired samples: Bone marrow cells can be analyzed along with peripheral blood using advanced flow cytometry methods that monitor the persistence of blasts and other leukemic subsets over time in these compartments. This type of analysis offers critical insight into the development of new therapeutics whether they are being evaluated in humanized mouse models or clinical trial participants. Flow cytometry continues to advance immuno-oncology research, especially for diseases involving the detection of rare cell populations. Consider working with preclinical and clinical flow cytometry experts to develop protocols for future immuno-oncology studies.
Enhancing CAR T Cell Therapy: Optimizing Preparation for Superior Results
Chimeric antigen receptor-mediated T cell (CAR T cell) therapies have revolutionized the treatment of hematologic malignancies and solid tumors. This therapy uses T cells, typically harvested from patients, that are engineered to express chimeric antigen receptors (CAR) specific to tumor cell antigens. CD19-targeting CAR T cell therapy was the first immunotherapy shown to effectively treat acute lymphoblastic leukemia[1], but a subset of patients relapse due to loss or poor engraftment of CAR T cells. Here we highlight advances in CAR T cell therapy to improve the quality of the immunotherapy product ex vivo for more effective responses in vivo. Culture Conditions T cells from peripheral blood mononuclear cells (PBMCs) are the primary cellular product used for CAR T cell therapy, but several steps must be carried out ex vivo to ensure that enough cells are made for therapeutic efficacy. The best treatment outcomes have been linked to high levels of CAR T cell engraftment and persistence upon transfer into a patient[2]. Ex vivo culture methods have been optimized to expand T cells, and cultures using less differentiated T cells, like stem cell memory T cells or naïve-like T cells, have been linked to better persistence upon transfer[3]. Recent studies are characterizing phenotypic features of T cells that preserve “stemness” upon ex vivo culture but still allow for expansion and expression of CAR T receptors. Reduced culture time has been one of the most effective methods for improving engraftment and antitumor responses but is limited by the number of cells yielded by this minimal manipulation process[4]. Biodistribution The successful engraftment and persistence of CAR T cells depend greatly on where cells migrate upon transfer into patients. Preserving stem cell-like features correlates with better engraftment, especially if donor-derived cells are unavailable and autologous cell sources must be used[5]. In vivo imaging studies have been very helpful in understanding CAR T cell dynamics and anti-tumor responses. Studies in mouse models have indicated that CAR T cells can get trapped in tissues, including the lungs, which can limit access to tumor targets[6]. Analysis of CAR T cells with tumor cells also revealed extensive functional heterogeneity, including CAR-T cells that can interact with tumor cells but not exert cytotoxic effector functions. A recent first-in-human study examined the biodistribution of radioisotope-labeled CAR T cells and confirmed that these cells rapidly distribute to tumor tissue but are taken up by the liver and spleen and can persist systemically for up to two weeks[7]. As new in vivo imaging studies are carried out, these insights will inform how CAR T cell products are made and delivered and are likely to improve treatment outcomes. Advances in ex vivo methods for CAR T cell preparation are already improving outcomes, and these methods are broadly applicable to donor-derived or autologous T cell products. In vivo imaging methods are also delineating characteristics of CAR T cells that improve tumor targeting or result in misdirected tissue homing. Future in vivo and ex vivo studies are well-poised to further advance CAR T cell applications.
Unlocking the Potential of CAR-T Therapy for Treatment of Multiple Myeloma
Multiple myeloma (MM) is the second most common hematological cancer[1] and results from a series of mutations that lead to malignant plasma cells in the bone marrow.[2] The survival of MM ranges from 5 to 7 years on average, although patients with high-risk MM, defined as inactive TP53 or 1q21 activation, have reduced survival of 2 years.[3, 4] Due to the complexity of this disease, MM remains incurable, however, the emergence of CAR-T (chimeric antigen receptor T-cell) therapy has provided hope for MM patients. Pathogenesis Under normal conditions, plasma cells secrete polyclonal protein or immunoglobulins comprised of one heavy chain and one light chain.[5] MM is derived from a series of mutations in plasma cells and clonal plasmacytes that instead produce monoclonal proteins (M-proteins) within the bone marrow (BM).[6] This disease typically progresses from monoclonal gammopathy of undetermined significance (MGUS) that later develops into smoldering multiple myeloma (SMM) – both of which are asymptomatic plasma cell disorders with no end-organ damage.[7] MGUS is defined as a lower percentage of M-protein (<3g/dl) while SMM is defined as M-protein concentration exceeding 3g/dl.[8] It is thought that initial mutation events seen in MGUS, and ultimately in MM, that lead to M-protein secretion occur during somatic hypermutation or class-switch recombination events, namely translocations involving IgH locus and IgL locus.[7] The results of these mutations form an abundance of dysfunctional plasma cells within the bone marrow leading to reduced immune system responses and organ damage. Therapeutics Standard of care treatment for newly diagnosed MM includes a cocktail of high-dose chemotherapy agents: bortezomib, lenalidomide, and dexamethasone.[9] These chemotherapeutic agents are effective at damaging tumor cell DNA as well as tumor microenvironment. Following this aggressive combination therapy, patients undergo allogeneic stem cell transplantation (ASCT) to restore blood cell production.[10, 11] Despite the success of these treatments, many MM patients experience graft-versus-host disease (GVHD) and/or relapse,[12] making it essential for new and innovative treatments to emerge. One treatment that has shown promise is chimeric antigen receptor T cell (CAR-T) therapy. CAR-T therapy is a type of immunotherapy that genetically modifies the patient's own T cells to target and kill cancer cells. [12, 13] In MM patients specifically, B cell maturation antigen (BCMA) targeted CAR-T therapy has been highly successful in newly diagnosed and relapsed MM patients. [13] Furthermore, clinical studies have found that CAR-T therapy has a 76% remission rate and conclude better 5-year patient outcomes.[14] Today there are two FDA-approved CART-T therapies for MM, Carvykti and Abecma, with others in development.[15] Despite successes, there are notable challenges with CAR-T therapy including cytokine release syndrome (CRS) and neurotoxicity, which may be managed with appropriate and timely interventions. [13] CAR-T therapy is also costly and requires specialized manufacturing facilities, limiting its availability to patients. Importantly, new treatments outside of CAR-T therapy are also in development with novel targets, namely Selinexor. Selinexor targets Chromosome Region Maintenance 1 (XP01), a transporter of nuclear proteins with a key role in cell cycle regulation and proliferation.[16] XP01 has been implicated in several solid tumors, such as lung cancer, and is overly expressed in MM. Overexpression of XP01 allows tumor suppressor proteins, such as Rb, p53, and p21, to be exported into the cytoplasm rendering them inactive and therefore increasing anti-apoptotic cell signaling.[16, 17] Selinexor covalently binds to the “cargo-binding” groove of XP01 which prevents the displacement of tumor suppressor proteins mentioned above.[17, 18] As a result of its pre-clinical and clinical trial success, the FDA approved Selinexor as a combination therapy with dexamethasone for patients with more than one prior therapy and at least 4 prior dexamethasone therapies.[19, 20] While MM continues to be a challenging cancer to treat, therapies such as CAR-T therapy and Selinexor provide MM patients with new hope for lasting remission. New clinical trials, such as NCT05177536 to test iberdomide maintenance therapy, are underway to explore the efficacy of new therapies and interventions to improve MM outcomes.
Benefits of Immuno-Oncology for Colorectal Cancer
Colorectal cancer (CRC) is a leading cause of cancer deaths globally. Advances in early detection have improved survival rates, but patients diagnosed with metastatic CRC still have stubbornly poor 5-year survival rates. Standard treatments for CRC include surgery, chemotherapy, and radiotherapy, but alternative immuno-oncology therapies are showing promising results in CRC patients. Here we highlight advances in immuno-oncology therapies that are being used to treat CRC patients or are being pursued in preclinical and clinical studies. CRC Subtypes Molecular genetic analysis of CRC tumors is a critical diagnostic tool for classifying and treating this cancer. CRC tumors are typically classified as mismatch repair-deficient/microsatellite instability-high (dMMR—MSI-H) tumors, which have a high overall mutation burden, or mismatch repair-proficient/microsatellite instability-low (pMMR—MSI-L) tumors, which have a lower mutation burden[1]. Defects in MMR are associated with the accumulation of mutations and are caused by defects in mismatch repair proteins, and these defects are typically detected by frame-shift mutations in DNA repeat regions known as microsatellites. Mutations in the BRAF oncogene, particularly the activating V600E mutation, comprise a distinct subset of CRC. BRAF is a component of the mitogen-activated protein kinase (MAPK) pathway that normally functions downstream of the epidermal growth factor receptor to regulate transcription of genes involved in cellular growth and survival, but the BRAF-V600E mutation results in constitutive activation of BRAF and uncontrolled cellular proliferation and tumor growth[2]. Immuno-Oncology Interventions Immuno-oncology approaches that target immune checkpoint blockade have proven effective for several cancers, including CRC. T cells can initiate effective anti-tumor responses under ideal conditions, but the immunosuppressive tumor microenvironment of some cancer types inhibits T cell activation, usually through engagement of immune checkpoint molecules like PD-1 and CTLA4. For more than a decade, the development and use of immune checkpoint inhibitors (ICIs) has transformed the treatment of melanoma[3,4] and non-small cell lung cancer[5,6]. The first FDA-approved treatments include a monoclonal antibody (mAb) that targets CTLA4 (ipilimumab) and two mAbs that target PD-1 (pembrolizumab and nivolumab). These early studies suggested that tumors with high mutation burdens respond well to ICI, which may be due in part to the generation and presentation of tumor neoantigens that are recognized as non-self and can be targeted by cytotoxic T cells[7]. CRC tumors with the dMMR—MSI-H signature have a high mutational burden and typically have a high level of CD4+ and CD8+ tumor-infiltrating lymphocytes (TILs). These cells have shown elevated expression of PD-1, PD-L1, and CTLA4, which suggests that they may respond well to ICIs[8]. Indeed, several recent clinical studies have shown improvements in overall survival and progression-free survival in patients treated with individual or combined ICIs that target PD-1. Of note, interim results from a phase II trial that enrolls patients with MMRd locally advanced rectal cancer to receive neoadjuvant dostarlimab, a PD-1 inhibitor, showed sustained complete response 1 year after the end of treatment in all patients[9]. Other current studies are evaluating next-generation PD-1 inhibitors combined with chemotherapy and/or other biologics such as an anti-VEGF and anti-EGFR mAb[10]. In contrast, patients with pMMR–MSI-L have shown poor responses to PD-1 or CTLA4 blockade alone or in combination, which has led to the development of trials that explore different treatment combinations, including inhibitors of the MAPK pathway or angiogenesis[11]. Promising results have been presented in a phase I/II clinical trial (NCT04017650) in which patients with MSI-L and BRAFV600E were treated with BRAF+EGFR inhibitors, to induce a transient MSI-H phenotype, plus anti-PD-1 antibody[12]. Next-Generation Treatments Several cutting-edge immuno-oncology therapies are being explored for the treatment of CRC. Beyond combinations of individual antibodies, researchers are engineering bispecific antibodies that bind to tumor cells and T cells simultaneously to enhance anti-tumor T cell responses. One such bispecific antibody, CEA-TCB, is being tested in phase I trials alone or in combination with anti-PD-L1 to treat metastatic CRC[13]. Another novel approach includes adoptive cell therapies like chimeric antigen receptor (CAR) T cells, which are T cells collected from the tumor tissue or peripheral blood of a patient and engineered to bind to tumor antigens and potentiate anti-tumor responses. Oncolytic virus and bacteria-based vaccines are also being studied as potential CRC treatments. Besides treatments designed on the tumor's intrinsic genetic background, much effort is being put into deciphering the tumor microenvironment, with a particular focus on its interplay with the gut microbiota to modulate inflammation and immune response, known to be involved in the metastatic onset. These studies are leading to the identification of inflammatory/immune signatures that inform the therapeutic agents to be used to target the pro-oncogenic microenvironment and reactivate the immune system[14]. The advancement of immuno-oncology is already transforming the treatment of CRC and will contribute to better outcomes for all types of CRC in the decades to come.
Immunohistochemistry: a Powerful Tool in Cancer Research
Immunohistochemistry (IHC) is a technique that originates in the early twentieth century but continues to be a valuable method that forms the backbone of molecular pathology. IHC is used for histological examination of tissues and specifically detects the presence of a molecule, such as a tumor antigen. IHC uses antibody-based labeling in which the primary antibody detects the target of interest and the secondary antibody detects the primary antibody which is linked to a molecule for microscopic visualization. Many different secondary antibody labeling modalities exist, including fluorescence, enzyme-mediated reactions and colloidal gold, and different labels are suited to specific microscopy platforms. Consider these five aspects of IHC as you implement this technique in preclinical cancer research: Quantitative measurements. IHC can be used as a qualitative measurement, but unlike many other visualization techniques, IHC can also be used as a quantitative measurement because antibodies that label specific parts of tissues or cells can be counted by a pathologist or with a computer-aided system. Developing robust validated quantitative IHC staining and visualization methods allows researchers to rely on the accuracy of this data. Customizable. IHC methods can be adapted to detect any cellular marker, given that a monoclonal antibody exists or can be made that specifically detects this marker. Validation of a new primary or secondary antibody also includes determining any off-target staining caused by these antibodies as this can be a critical determinant in the utility of an antibody. The ability to customize IHC in this manner is crucial to preclinical research that seeks to identify new biomarkers associated with tumor progression or immunotherapy efficacy. Flexibility. IHC can be used on almost any tissue type so long as it is processed correctly. Tissue samples from model animals as well as clinical biopsies can be fixed and sectioned in advance and stained at later times. Tumor microarrays (TMAs) can also be created and stained for evaluation of novel tumor markers or screening efficacy of drug candidates. This flexibility in sample type and handling highlights the overall utility of IHC. Automation. Currently, several different systems exist for automated IHC staining, and advances in digital analysis of IHC samples have allowed larger batches of clinical samples to be processed and evaluated. Comparison of automated IHC methods with manual methods has determined that these new approaches are accurate, sensitive, and reproducible. The big picture. Several methods exist for staining different targets in an IHC sample, and this allows scientists and clinicians to gain critical insights into which cells and molecules are present in the tumor microenvironment (TME), including levels of immune checkpoint molecules or infiltration of critical anti-tumor cell types. Consider how IHC can complement other techniques that look at the tumor microenvironment, including flow cytometry and RNAseq. IHC will continue to be a powerful tool in preclinical and clinical cancer research. Consider revisiting this classic technique as it has matured with the technological advances of the 21st century. Champions Oncology’s histology and immunohistochemistry services are custom developed and fully optimized to meet your needs in preclinical research. With industry leading pathology expertise and innovative automated technology, Champions provides you with the highest quality endpoints for your in vivo and ex vivo studies.

Uncovering Advantages and Disadvantages of Ex Vivo Culture in Preclinical Cancer Research
Ex vivo culture systems have been instrumental to preclinical oncology research because they enable researchers to study basic features of tumor cells and carry out large-scale screens of drugs or biologics. Ex vivo cultures do not fully recapitulate physiological conditions, although some advances have been made by developing three-dimensional (3D) culture methods. Here we highlight the advantages and disadvantages of using ex vivo cultures for preclinical oncology research. Cell culture: simple, inexpensive, but imperfect Ex vivo cell cultures of tumors were first developed as adherent two-dimensional (2D) monolayers that were grown in culture flasks or flat dishes. This approach is useful for studying tumors' basic cell biology and carrying out drug screens and preliminary toxicology and pharmacokinetic studies[1]. 2D cultures can be developed rapidly and cultured over longer periods of time using readily available tissue culture reagents. Unfortunately, 2D culture systems are limited in their applications because they do not accurately model the tumor microenvironment, and tumor cell morphology and function change under these culture conditions. Over the last several decades, 3D cultures have advanced significantly[2]. Several different systems now exist for 3D cultures, including liquid suspension cultures passaged on non-adherent plates, cultures grown in semi-solid substrates like soft agar or Matrigel, and cells grown on scaffolds like collagen. 3D culture systems better mimic characteristics of the tumor microenvironment, including cell-cell interactions, hypoxia, and reduced sensitivity to drug treatment[3]. (Visit our dedicated webpage to learn more about Champions' 3D tumor model offer) 3D cultures for drug discovery have become widely adopted over 2D models because they better predict in vivo efficacy and metabolic responses to drug treatments. A major advance in 3D tumor culture systems is the development of 3D spheroid and organoid cultures[4]. Spheroids are clusters of cells derived from tumors or other complex tissues and they cluster together through cell-cell adhesion. Organoids are more complex cell clusters derived from stem cells and can self-assemble and regenerate to form a smaller version of the original tumor or organ tissue. Tumor spheroids are widely used for drug screening and cytotoxicity assays and can be formed from dissociated tumor tissue or circulating tumor cells[5]. (Read our blog "Using 3D Ex Vivo Tumor Models for Oncology Research: An Expert Guide") Tumor organoids are even more accurate reflections of tumors growing under physiological conditions and are now widely used to compare the efficacy of standard-of-care treatments versus targeted therapies in addition to their use in drug screens[6]. Spheroids and organoids can undergo genomic editing as well, and this approach has been instrumental in identifying mutations that drive tumor growth or cause drug resistance[7-8]. (Read our blog "A Step-by-Step Guide to Design a 3D Tumor Model Co-Culture Study") Ex vivo + in vivo = the most effective strategy Animal models, especially patient-derived xenograft mouse models, are a critical bridge between preclinical studies and clinical trials. However, due to the lower costs and the compatibility with high throughput analysis, ex vivo tumor cultures are still the preferred method for ranking therapeutic agents, determining drug combination regimens, investigating mechanisms of action, and validating targets. Additionally, ex vivo tumor cultures are essential when studying certain hematological tumors, which are difficult to engraft and cannot be passaged in vivo because of their “liquid” nature. Both ex vivo and in vivo studies continue to work together to advance oncology research, but each year brings new advances in ex vivo culture systems that enhance their overall impact in predicting treatment responses.
Harnessing the Power of Oncolytic Viruses in the Fight Against Cancer
We typically consider viruses as infectious agents or vaccine vectors, which are non-replicating entities that express vaccine antigens. More recently, researchers have been working to develop oncolytic viruses, which are engineered to specifically infect and replicate in tumor cells. This targeted infection results in tumor cell lysis, activation of anti-tumor immune responses, and the release of new oncolytic virus particles that can infect other tumor cells. The concept of oncolytic viruses emerged in the early 20th century when scientists observed that a leukemia patient spontaneously went into remission for a brief time following an influenza virus infection. Early attempts to develop non-specific oncolytic viruses were plagued with complications related to poor efficacy and safety issues. As our understanding of targeted immunotherapy for the treatment of tumors has evolved, oncolytic viruses are gaining traction again and an oncolytic herpesvirus (talimogene laherparepvec or T-VEC) engineered to target metastatic melanoma was approved for clinical use by the FDA in 2015 [1]. Advantages of Oncolytic Viruses Oncolytic virus development has expanded significantly in the last two decades. Some of the most widely used viruses that are engineered to target tumors include adenoviruses, alphaviruses, herpes simplex viruses (HSV), rhabdoviruses, and vaccinia viruses. Viruses can be engineered to target tumor cells for lysis, such as replication-competent HSV-1 deletion mutants (e.g. thymidine kinase or ribonucleotide reductase mutants) that can only replicate in rapidly dividing tumor cells. Similarly, T-VEC is a modified HSV-1 virus with a gene deletion in ICP34.5, which allows for antiviral responses by normal cells but not tumor cells, thus allowing for tumor-specific virus-mediated destruction [2]. Oncolytic viruses can be modified to express immune molecules (e.g. TNF, IL-12, or chemokines) that promote proinflammatory responses and increase recruitment of macrophages and T cells for enhanced antitumor immunity in addition to oncolytic activity [3-4]. Oncolytic viruses can also induce apoptosis of tumor cells or enhance the uptake of chemotherapeutic agents [5]. Currently under pre-clinical investigation is a vaccinia virus carrying a TGFβRII inhibitor that has proved effective in causing tumor regression in mouse tumor models and shown an even greater effect when combined with checkpoint inhibitor therapy [6]. This approach overcomes the difficulties of targeting TGFβ, limiting side effects due to the targeting of non-tumor cells. Another promising approach currently being evaluated pre-clinically is a therapy using CAR-T and TCR-T cells infected with myxoma virus. This approach induces autosis and adaptive immunity in mouse models of tumors to restrain antigen escape [7]. Some oncolytic viruses can induce long-term immunity to tumors and prevent metastasis or the re-occurrence of these cancers, thus making them attractive therapeutic candidates. Clinical trials using oncolytic viruses are currently ongoing for numerous solid tumor types including glioblastoma, breast cancer, lung cancer, and bladder cancer [8]. Evaluating Oncolytic Viruses Oncolytic viruses are typically grown in tissue culture systems and are evaluated in vitro using a panel of tumor cell lines, which provides insight into tumor specificity and mode of action. These modified viruses can then be tested in a wide range of animal models, including immunocompetent mice, such as those used for syngeneic mouse tumor models, and immunocompromised mice, which include humanized mice that carry patient-derived tumor xenografts. Certain oncolytic viruses, such as vaccinia virus, require that researchers be vaccinated against this virus before laboratory handling, but in most cases, these viruses can be handled under BSL-2 conditions. Conclusions Despite great success in preclinical studies, translating oncolytic virus therapy to the clinic can be quite challenging. One major obstacle is the route of administration, with the oncolytic viruses needing to be injected directly into the tumor. While this can be simpler for superficial tumors like melanoma, it becomes much more complex to reach tumors that grow in deeper body organs. Many different varieties of oncolytic viruses are currently being evaluated preclinically or in clinical trials and the next decade promises further advances in this cutting-edge field.
Ultimate Guide: Designing a Mouse Clinical Trial & Data Analysis
Cancer is a complex disease, and developing novel, effective treatments requires testing in preclinical models. Mouse clinical trials represent a critical step in translating promising anti-cancer therapies from bench to bedside. When combined with advanced bioinformatics and analytics tools, mouse clinical trials are a powerful experimental approach to identifying target patient populations, planning cohort extensions, selecting the right combination drug, elucidating the mechanism of resistance, or identifying translational biomarkers [1]. However, conducting a successful mouse clinical trial requires expertise in designing the study, analyzing the data, and interpreting the results. In this blog post, we will guide you through the essential steps of designing a mouse clinical trial and analyzing the resulting data. This guide includes tips on powering the study, preparing the dataset, grouping responders vs non-responders, identifying biomarkers, and integrating proteomics in a multiomics model. Step 1: Powering the study In a mouse clinical trial, animals implanted with PDX models are surrogates of the patients whom PDX models were derived from. Before starting a mouse clinical trial, it is essential to determine the sample size required to achieve reliable and statistically significant results. It is important to enroll a large enough number of PDX models (which corresponds to the number of patients we would enroll) and enough animals per PDX model (so as not to lose a patient if we lose a mouse). Power analysis will estimate the minimum number of animals needed to detect a difference in tumor growth inhibition (TGI) between the treatment and control groups. Typically, researchers set the power at 80% and α at 0.05, which means that there is an 80% probability of detecting a significant difference between the two groups if one exists. It is also recommended to power mouse clinical trials according to the study goal and endpoint. For instance, a survival endpoint may require larger group sizes than a TGI endpoint. If the goal of the mouse clinical trial is to identify a novel biomarker of response, a larger number of models will need to be included in the design. Step 2: Select models Model selection is a crucial step when designing a mouse clinical trial and needs to be done by taking into account several parameters associated with the clinical history and molecular characteristics of the models so that the PDX model panel recapitulates the targeted patient population. Tumor indication and/or mechanism of action of the drug are usually the two key parameters that guide PDX model enrollment in a mouse clinical trial, and the deeper the information in terms of clinical classification and molecular profiling the more representative will be the PDX model panel [2]. (Read our blog: "How to Use Metadata as Your Model Selection GPS") Step 3: Preparing the dataset Once the study is completed, the next step is to prepare the dataset for analysis. This includes checking for outliers and removing/excluding missing data. One critical factor in analyzing mouse clinical trial data is the identification of PDX models with common response profiles. This is usually evaluated by a modified version of the clinical Response Evaluation Criteria in Solid Tumors (mRECIST). The response rate can be calculated by comparing the tumor volume at day 0 either with the volume corresponding to the best response measured on treatment or with the tumor volume measured on the final day of treatment. The threshold value can vary depending on the tumor type and drug mechanism of action. Therefore, it is crucial to validate the threshold on a panel of known drugs. Step 4: Grouping responders vs non-responders Once the threshold is established, the next step is grouping responders vs non-responders. Responders are mice that show a reduction in tumor volume or mass greater than or equal to the critical value, and non-responders are mice that show tumor reduction below the critical value. These groups are used to compare gene expression profiles and identify potential biomarkers of response [3]. Step 5: Identifying biomarkers Bioinformatics analysis can be used to identify biomarkers of response. One widely used approach is differential gene expression analysis (DGEA), which compares the gene expression profiles of responders and non-responders to identify genes that are differentially expressed. Another approach is differential gene set enrichment analysis (DGSE), which identifies functional gene sets that are enriched in the responders and non-responders. Partial least-squares (PLS) regression, a dimensional reduction method part of DIABLO multi-omics integration workflow [4], is commonly used to identify the most influential genes within a multi-omics gene network that can predict response. It is very important that mouse clinical trials are designed accordingly when performing multi-omics analysis as an endpoint. (Watch our webinar: "Identify MOA and Companion Biomarkers in Oncology using Multi-Omic Analyses") Step 6: Integrating proteomics for improved target validation and biomarker identification Proteomics approaches, such as mass spectrometry-based proteomics, can provide quantitative information on protein expression levels and post-translational modifications. Given the lack of correlation between protein abundance and RNA expression, integration of proteomics and phospho-proteomics data in a multi-omics model can greatly improve the accuracy of therapeutic target expression for model selection, provide a more comprehensive understanding of the molecular mechanisms underlying drug response, and provide an important additional molecular annotation for biomarker identification via single or multi-omics analysis [5]. (Read our blog: "4D Proteomics: Adding Dimension to Protein Detection") In this blog post, we have provided a step-by-step guide on how to design and analyze a mouse clinical trial, and shown how a successful mouse clinical trial requires careful planning and expertise in bioinformatics. Lumin Acuity offers tailored bioinformatics and computational biology solutions to support your mouse clinical trial planning and data analysis, accelerating decision-making and driving actionable results. Well-designed and executed mouse clinical trials can be instrumental in developing effective cancer therapies and improving patient outcomes.
Advancing Therapeutic Strategies for Myelodysplastic Syndrome
Myelodysplastic syndromes (MDS) are a group of hematological malignancies that manifest in hematopoietic stem cells and are caused by ineffective hematopoiesis.[1] Currently, MDS is defined as unexplained cytopenia combined with abnormalities in cell maturation leading to dysplastic features in >20% of myeloid cells.[2] MDS is one of the most frequently diagnosed malignancies in the United States and progresses to acute myeloid leukemia (AML) in 30% of patients.[2] While the initiation of primary MDS is not well understood, research suggests that somatic DNA injury, defective DNA repair, impaired immunological responses, and dysfunctional cell signaling play an important role in early stage MDS development.[3] Like other malignancies, MDS is positively selected for through gene mutations[4] with the average MDS patient carrying nine somatic mutations, such as TET2, TP53, and RUNX1.[4, 5] These mutations may not directly drive MDS, however, evidence indicates that mutations to genes involved with DNA methylation and RNA splicing greatly contribute to dysregulation of genes critical to hematopoiesis, such as GATA1, KLF1, and HOXA9.[6] MDS is classically characterized as hematopoietic stem cells (HSC) with clonal advantages due to somatic mutations or cellular dysfunction, discussed above. Studies have also identified the critical role of bone marrow microenvironments (BME) in MDS progression, particularly cytokine alterations and activities. Across several studies, high serum levels of TNF-α, TGF-β, IL-6, and IL-8[7] are present in MDS patient bone marrow (BM).[8] These proinflammatory molecules are typically associated with higher apoptotic rates, and in MDS patients, molecules such as TNF- α, correlate to poor MDS treatment performance.[7] Furthermore, studies have also identified the critical role of malignant clones in MDS pathogenesis. Mesenchymal stromal cells (MSCs) are a key component of the bone marrow that regulate hematopoiesis and have immunomodulatory properties. In contrast, dysplastic MSCs in the bone marrow can create a microenvironment that supports clonal expansion of malignant cells. Dysplastic MSCs from MDS or AML patients have phenotypic abnormalities, including aberration in secreted proteins and cell surface protein expression, as well as increased senescence and decreased survival.[9,10] Disrupted methylation profiles are observed in both MDS malignant clones and MDS-MSCs. These methylation changes impact multiple signaling pathways which create a chronically inflamed BME that is harmful to normal HSCs and supports the expansion of MDS clones.[11] While less understood, de novo MDS has been linked to chemotherapy, radiotherapy, and environmental carcinogens like benzene.[12] Therapy-related MDS (tMDS) is prevalent in 10-20% of patients 20 years after chemotherapy and/or radiation, and the World Health Organization (WHO) recognizes alkylating agents, like topoisomerase II inhibitors, as initiators of tMDS.[12] Due to the high incidence of MDS in elderly populations[13] with co-morbidities, treatment by bone marrow transplantation is often contraindicated and few therapeutic options exist. Patients with MDS are typically divided into two different categories: low-risk MDS and high-risk MDS. The division of MDS into these two groups is useful for researchers and clinicians developing therapies for this highly heterogeneic disease.[14] Low-risk MDS therapies target symptoms of cytopenia through the use of erythropoiesis stimulating agents (ESA), such as epoetin alfa and darbepoetin alfa.[15] The Food and Drug Administration (FDA) has also approved two hypomethylating agents (HMAs), 5-azacitidine and decitabine, for use in patients with low-risk and high-risk MDS. While both standards of care noncompetitively inhibit DNA methyltransferase (DNMT1) and promote hypomethylation of DNA to block DNA synthesis,[16] 5-azacitidine integrates with RNA and interferes with ribosomal assembly to also limit tumor protein synthesis.[16, 17] In 2023, the FDA and European Medicines Agency (EMA) also approved luspatercept, a recombinant fusion protein that enhances late-stage erythroblast differentiation, for low-risk MDS. Luspatercept indirectly moderates ineffective erythropoiesis by interacting with broad spectrum inhibitory signals, namely transforming growth factor-β (TGF- β) superfamily signaling.[18, 19] Downstream, signaling molecules, SMAD2 and SMAD3, negatively regulate TGF- β. Luspatercept works by binding to activin receptor type IIB on TGF- β which disrupts SMAD2 and SMAD3 signaling therefore improving erythropoiesis and boosting RBC production in MDS patients.[20, 21] As our understanding of MDS pathophysiology improves, more effective treatment options will become available. Researchers are actively evaluating the toxicity of allogeneic hematopoietic stem cell transplantation, a therapy with curative potential.[19] Given the nature of such treatment which involves conditioning chemotherapy, the risk of aplasia and other graft-versus-host disease need to be adequately assessed. Additional studies, such as a newly announced Montefiore Einstein Comprehensive Cancer Center clinical trial,[22] are investigating new therapies to develop novel therapeutic strategies.
A Multi-Omics Driven Approach for Advancements in Pancreatic Cancer
Pancreatic ductal adenocarcinoma (PDAC) is one of the most common and aggressive forms of pancreatic cancer that has remained difficult to diagnose early and treat successfully. PDAC has a five-year survival rate below 10% and is one of the leading causes of cancer death. Complete surgical resection is one of the few curative treatment modalities, and chemotherapy protocols have limited efficacy[1]. Unfortunately, most existing immunotherapy-based treatments are also associated with poor response rates[2]. Recent analyses of PDAC samples have provided critical insights into genetic alterations that drive tumorigenesis and have identified potential therapeutic targets. Here we highlight several of these key findings and how they may lead to advances in PDAC treatment. PDAC arises in the epithelial cells of pancreatic duct, or ductules, and is thought to progress in a manner like other carcinomas, in which the normal epithelial cells transition into pre-invasive pancreatic intraepithelial neoplasia lesions that eventually form invasive PDAC[3]. Most PDAC tumors carry somatic mutations in oncogenes, particularly KRAS, TP53, CDKN2A, and SMAD4[4]. KRAS mutation is the most frequent event in PDAC. The assumption that KRAS is an undruggable target based on extensive drug screens that showed in vitro inhibition but limited or no efficacy in animal models[5] is finally being challenged by the very promising results obtained by administration of Sotoresib (KRAS p.G12C inhibitor) in patients with advanced pancreatic cancer (NCT03600883), and by the identification of small-molecules that inhibit KRAS p.G12D in preclinical studies[6,7]. The need for druggable targets or biomarkers for PDAC has led to more innovative approaches to identify unique molecule attributes. A recent comprehensive proteogenomic characterization of PDAC pancreatic ductal tissues compared against paired normal adjacent tissue, and the findings validated known mutations in oncogenes[8]. This study also defined previously unknown genomic alterations and analyzed differences in protein expression and protein phosphorylation status between tissues. This comprehensive analysis identified a panel of proteins linked to early stage PDAC, and phosphoproteomic analysis identified several signaling pathways downstream of KRAS, including PI3K/AKT/mTOR and MAPK/ERK, that may be targeted by existing kinase inhibitors. The PAK1/PAK2 kinases were also identified as dysregulated in PDAC tissues and have the potential to be targeted therapeutically. Recent studies have also better characterized the tumor microenvironment (TME) of PDAC and how this may limit treatment efficacy[9]. PDAC tumors tend to be highly heterogenous with a dense stroma and disorganized blood vessels, which impair drug penetration. The TME is enriched for myeloid derived suppressor cells and regulatory T cells and displays a low mutational burden, thus making this a “cold” tumor immunophenotype that is resistant to existing immunotherapies. Current studies are examining immunotherapy or drug-based strategies to turn “cold” PDAC tumors into “hot” tumors that would be receptive to immune checkpoint blockade. Further advances in PDAC research will require similar extensive proteogenomic studies that better define dysregulated pathways that trigger early events in tumor formation and may function as early biomarkers for disease. The search for novel therapeutic targets or combinations of targets is also essential to improving the currently dismal array of treatment options for PDAC patients. Champions Oncology has 81 highly characterized Pancreatic Cancer Models available for preclinical studies, click below to learn more about how these models can propel your research.
4D Proteomics: Adding Dimension to Protein Detection
The field of oncology research has evolved significantly over the years, with new technologies constantly emerging to improve our understanding and treatment of cancer. One such technology is 4D proteomics, which utilizes four dimensions (retention time, mass, intensity, and ion mobility) to improve the detection of low-abundant peptides in complex biological samples. This powerful technique has proven to be particularly useful in personalized medicine and biomarker discovery for cancer research[1]. What is 4D Proteomics? Proteomics is the study of all proteins present in a given sample or organism. Traditional proteomic methods have typically focused on identifying and quantifying proteins in a sample based on their mass and abundance. However, this approach often falls short when it comes to detecting low-abundant peptides, limiting our ability to fully understand the complexity of biological systems. That's where 4D proteomics comes in. This technique adds an additional dimension (ion mobility) to the traditional three dimensions of mass, retention time, and intensity. Ion mobility measures the ability of a molecule to move through a buffer gas under an electric field, providing another valuable data point to improve peptide separation and identification[1]. The Benefits of 4D Proteomics in Oncology Research The use of 4D proteomics has greatly improved the sensitivity and specificity of peptide identification, making it particularly useful in oncology research where small changes in protein levels can have significant implications. The addition of ion mobility as a fourth dimension has also improved peptide separation and identification, making it easier to analyze complex samples such as blood or tissue from cancer patients. This is especially crucial in personalized medicine, where individualized treatments are tailored to a patient's specific cancer and its unique molecular characteristics[2]. Additionally, ion mobility helps differentiate each phosphopeptide isoform, accurately identifying the phosphorylation site on the peptide and quantitating each peptide form. This is critical for example in measuring certain kinases whose activity or substrate selectivity is controlled by phosphorylation at specific sites[3]. Conclusion In conclusion, the use of 4D proteomics has revolutionized the field of oncology research by improving our ability to detect low-abundant peptides and analyze complex samples. This technology has proven to be particularly valuable in personalized medicine and biomarker discovery for cancer research. As more studies continue to utilize 4D proteomics, we can expect further advancements in our fight against cancer.

Recent Developments in Targeted Therapy for Non-Small Cell Lung Cancer
Lung cancer is an aggressive malignancy that correspond to approximately 12% of newly diagnosed cases per year and that remains one of the leading causes of cancer-related deaths (21%)[1]. Non-small cell lung cancer (NSCLC) is the most common form of lung cancer and is typically diagnosed at advanced stages[2]. Extensive genomic studies of NSCLC have been essential to identifying mutations that drive oncogenesis and has been critical to the development of targeted therapies. Here we highlight progress in NSCLC treatment and identify areas of ongoing research. Chemotherapy For decades, chemotherapy was the primary treatment for NSCLC, and platinum-based chemotherapies are still used today in combination with radiotherapy and targeted therapies[3]. Platinum-based chemotherapies have been improved since their initial development in the late 20th century, and “platinum doublets” or “triplets”, which combine two or three different platinum-based cytotoxic compounds, have been shown to better target NSCLC and cause less toxicity[4]. Chemotherapy is still used as a first line treatment for advanced NSCLC, but overall effectiveness is limited when used alone. Advances in targeted therapies and immune checkpoint blockade have greatly enhanced the effectiveness of chemotherapy on overall survival and progression-free survival[5]. Targeted Therapies Genetic analysis of NSCLC tumors has led to the use of targeted therapies. Mutations in the epidermal growth factor receptor (EGFR) were identified as the most prevalent defect in NSCLC patients across genders, ethnic groups, and smoking status[6]. Several common mutations have been characterized, but numerous rare mutations have also been identified, and most of these mutations target the tyrosine kinase domain, which results in unchecked cellular proliferation due to engagement of downstream MAKP, PI3K, and STAT signaling pathways. First (gefitinib and erlotinib) and second (afatinib and dacomitinib) generation tyrosine kinase inhibitors (TKIs) have been approved by the FDA to treat EGFR-mutated NSCLC. Unfortunately, treatment resistance and disease recurrence occur at a very high rate, and these TKIs are also associated with significant toxicity and undesirable side effects that result in adjustments of treatment regimens[5]. Third generation TKIs (e.g. osimertinib) that target specific EGFR mutations associated with acquired resistance to first and second generation TKIs have been approved and have been very successful in treating EGFR-mutated NSCLC as a first or a second line treatment. Because resistance to third generation TKIs eventually also arises, fourth generation TKIs are currently under development or are being used in ongoing clinical trials. Many of these next generation inhibitors have been carefully designed to optimize target specificity and reduce adverse events frequency and severity[7]. Mutations in other genes have also been observed at low frequency in NSCLC, and several clinical trials are now evaluating targeted therapies with inhibitors that target these genes, including, ALK, MET, ROS1, and BRAF among others[7, 8]. Capmatinib, an inhibitor targeting METex14 mutation, and adagrasib and sorotasib, both targeting KRAS G12C mutation, have recently been granted approval for treatment of NSCLC tumors presenting with the corresponding mutation[9, 10, 11]. In addition to small molecule inhibitors, Antibody-Drug Conjugates (ADC), a class of anti-cancer therapy capable of transporting cytotoxic drugs directly to tumor cells, are currently developed against NSCLC antigenic targets[12]. Immunotherapy Immunotherapies that target the immune checkpoint molecules like Programmed Death (PD)-1 or its ligand PD-L1 have been shown to have therapeutic effects as second-line treatments in combination with chemotherapy[13] for patients lacking mutations that can be treated with approved targeted therapies. While several of these trials have shown improvements in overall survival, serious adverse events have also been elevated. In addition to immune checkpoint targeting, other immunotherapies are currently under development, and a bispecific antibody has been recently approved for the treatment of a subset of NSCLC patients[14]. Biomarkers The availability of oncogenic driver mutations and of immune checkpoint targeted therapies have revolutionized NSCLC treatment and require the concomitant development of diagnostic tools to stratify patients and maximize the efficacy of these therapeutic options [15]. Oncogenic mutations are usually good companion biomarkers to indicate a patient as eligible to receive the associated treatment, however, diagnostic strategies need to be implemented to monitor patient response and anticipate or detect acquired resistance as early as possible. Immune checkpoint proteins are also currently tested as biomarkers to stratify patient, as an example a PD-L1 diagnostic test has been recently approved by FDA as companion biomarker to identify patients who are candidates for adjuvant treatment with atezolizumab [16]. Conclusion Advances in NSCLC have shifted the median overall survival from 2-4 months in the 1960s to more than 2 years since advent chemotherapy combined with immunotherapy or targeted inhibitors in the 2010s. Our deeper understanding of mutations that drive NSCLC and our ever-expanding arsenal of targeted therapies or immune modulators will further improve survival and quality of life for NSCLC patients.
Potentials of BTK Therapies for Chronic Lymphocytic Leukemia
Chronic lymphocytic leukemia (CLL) is one of the most common forms of adult leukemia, and its chronic nature has made it a challenging blood cancer to completely cure. CLL affects B cells and is typically classified into two categories: little or no somatic hypermutation in the immunoglobulin heavy chain variable region (IGHV), called unmutated CLL, or high mutation levels in the IGHV gene, called mutated CLL.[1] Unmutated CLL is more aggressive than mutated CLL, and the presence of these abnormal IGHV sequences, a part of B cell receptors (BCR), leads to abnormal BCR signaling and the uncontrolled proliferation of leukemic cells. Bruton’s tyrosine kinase (BTK) is an essential enzyme downstream from the BCR and is responsible for propagating the signaling cascade initiated by BCR engagement with pathogen-associated antigens.[2] Notably, BTK has been shown to be constitutively active in CLL and drives both leukemic cell proliferation and lymph node homing.[3] As antigen binds to the BCR, multiple protein tyrosine kinases are activated via interactions with the cytoplasmic domain of the BCR, including Lyn and Syk, as well as translocation of BTK to the plasma membrane through interactions with phosphatidylinositol-3,4,5 (PIP3).[4] Lyn and Syk phosphorylate BTK to activate multiple non-receptor protein tyrosine kinase signaling pathways, including NF-κB, MAPK, and phospholipase C gamma (PLCγ). Under normal conditions, this pathway leads to controlled B cell proliferation and differentiation, however, this same pathway leads to uncontrolled B cell proliferation in malignancies, such as CLL. The role of BTK in CLL has drawn researchers to develop BTK inhibitors, such as ibrutinib - an FDA (Food and Drug Administration)[5] and EMA (European Medicines Agency)[6] - approved CLL treatment. Ibrutinib forms a covalent bond with a cysteine residue (C481) at BTK’s active site, and thus inhibits kinase activity, including autoactivation.[3] This kinase inhibition shuts down BCR signaling, reduces B cell proliferation, and promotes apoptosis of leukemic cells. Ibrutinib is effective as a standalone treatment for mutated and unmutated CLL and when combined with rituximab is an effective therapy for relapsed CLL.[1] In general, ibrutinib is well tolerated and shows continued efficacy during extended treatment periods.[7] While ibrutinib has been a clinical success, there are a subset of CLL patients who develop resistance to this therapeutic. This resistance has been mapped to a C481S mutation that prevents ibrutinib from covalently binding to BTK resulting in continuous BCR signaling within leukemic cells.[3] Next-generation, reversible BTK inhibitors, such as ARQ 531, are currently being developed for relapsed/refractory CLL. ARQ-531 is a non-selective BTK inhibitor that suppresses signaling in cells with C481S BTK and PLCγ mutations. ARQ-531 also has an additional inhibitory activity against ERK signaling.[8] Furthermore, the FDA has recently approved LOXO-305, a non-covalent, reversible BTK inhibitor. LOXO-305 inhibits signaling in cells with wild-type or C481S-mutated BTK.[9, 10] In contrast, acalabrutinib, a second-generation irreversible BTK inhibitor has also been approved by the FDA. Acalabrutinib has a shorter half-life, allowing for variable dosages, and has increased C481 specificity that enhances its BTK inhibitory effects. The biochemical properties of acalabrutinib make it a suitable option for patients with treatment naïve CLL.[11] In January 2023, the FDA and EMA approved an additional second-generation irreversible BTK inhibitor, zanubrutinib, for CLL.[12] Zanubrutinib’s design was guided by a structure-activity strategy to generate sustained BTK occupancy. Zanubrutinib exhibits reduced ITK and EGFR inhibition and has 4x longer half-life than acalabrutinib.[13] As such, it persists at high concentrations within the body making it available to re-inhibit newly synthesized BTK proteins, a unique difference from ibrutinib and acalabrutinib.[14] In a phase I/II study, all CLL patients treated with zanubrutinib had complete and sustained BTK occupancy in peripheral blood mononuclear cells and lymph nodes.[15] Zanubrutinib’s performance supports the hypothesis that increased BTK selectivity maximizes BTK inhibition and drug efficacy. Additionally, zanubrutinib’s enhanced BTK specificity reduces the incidence of off-target toxicities which may reduce side effects commonly associated with ibrutinib, such as cardiac events, subdural hematomas, and gastrointestinal bleeding.[10, 16, 17] This is particularly relevant to CLL as this class of BTK inhibitors are used by CLL patients indefinitely. BTK inhibitors have become an invaluable CLL treatment and will continue to improve and provide therapeutic benefits. Additional studies such as NCT03734016 are underway to evaluate the effects of zanubrutinib and BTK inhibitors with other targeted therapies, which may improve the efficacy and durability of these treatments.
The Complex Case of Hepatocellular Carcinoma Clinical Management
Hepatocellular carcinoma (HCC) is one of the most prevalent types of liver cancer, and it usually develops in patients with underlying cirrhosis or chronic liver disease and metabolic syndrome.[1] HCC can spread to other body parts and affect patients' survival rates and outcomes severely. In this blog post, we will provide essential insights into HCC metastases and delve into the therapeutic options. Over 60% of patients with HCC are diagnosed in advanced stages, leading to an overall 5-year survival rate of 21%. However, this rate is between 60% and 70% in patients with early diagnosis who undergo liver transplants.[2] Patients diagnosed with advanced-stage HCC, defined by the presence of vascular invasion or extrahepatic metastases, have a poor prognosis with a median survival of less than 1 year. [3] Early detection of HCC is vital in increasing therapeutic opportunities and improving patients’ outcomes, and new diagnostic biomarkers are under investigation to enhance HCC screening.[4] Available treatments for HCC metastases depend on the tumor's location, the extent of the cancer, and the patient's general condition. Surgical resection, liver transplant, radiofrequency, and microwave ablation are all first-line treatments for tumors that have not spread to other body parts. Chemoembolization is commonly used and improves survival in asymptomatic patients with multifocal regional disease. [5] Systemic therapies, such as sorafenib and lenvatinib have been shown to be effective in prolonging survival rates and outcomes in patients with advanced HCC. These agents are multikinase inhibitors that block tyrosine kinases involved in angiogenesis, cancer development and growth, and tumor microenvironment regulation. They are used as first-line therapy in advanced HCC with contraindications to VEGF and immune checkpoint inhibitors. Immune checkpoint inhibitors are instead used together with bevacizumab in patients without contraindication. Alternative tyrosine kinase inhibitors, such as regorafenib, cabozantinib, and ramucirumab are used as second-line treatments.[6] Scientists are also researching new therapies for advanced HCC, including novel molecular-targeted therapies (such as STAT3 and CDK4 inhibitors) and immunotherapies to use as single agents or in combination. Several clinical trials are currently underway to investigate the efficacy and safety of these new therapies for advanced and metastatic HCC.[7] Hepatocellular carcinoma is a critical concern for clinicians and patients, given the cancer progression and severity. Validating new diagnostic biomarkers and developing new therapeutic approaches to overcome drug resistance will be paramount in improving patients’ survival and quality of life.
Hitting the Target: Latest Developments in Anti-Her2 Therapy
Human epidermal growth factor receptor 2 (HER2; ERBB2) has been shown to induce oncogenesis in several cancers, particularly breast and gastric cancers. HER2 is estimated to be overexpressed in ~20% of all breast cancers, and outcomes for these breast cancers were poor before HER2-targeted therapies[1]. Our understanding of mechanisms by which HER2 drives tumor growth and metastasis led to one of the first targeted therapies for treating HER2+ breast cancers. Here we highlight what is known about HER2 and oncogenesis. and how this information can be applied to the development of new targeted therapies. Gene amplification of the ERRB2 oncogene is one of the most common genetic defects detected in HER2+ tumors and typically causes overexpression of HER2 at the cell membrane[2]. HER2 proteins more readily dimerize with other HER2 proteins to form homodimers or with other ERRB members to form heterodimers. These complexes induce oncogenic signaling cascades, including MAPK and PI3K/AKT/mTOR, which cause cellular proliferation, angiogenesis, and resistance to apoptosis[3]. Due to the dominant role of HER2 in breast cancer, anti-HER2 antibodies were developed as one of the first targeted therapies. In 2006, clinical trial results of breast cancer patients treated with the anti-HER2 antibody trastuzumab showed significant improvements in progression-free survival and overall survival[4],[5]. Unfortunately, ~20-25% of metastatic breast cancer patients develop resistance to anti-HER2 therapies. More recent studies have focused on engineering new forms of anti-HER2 antibodies or developing HER1/2 inhibitors that can be used alone or in combination with other targeted therapies. The current standard of care for HER2+ breast cancer includes chemotherapy with adjuvant HER2-targeted therapies and endocrine therapy as indicated. Advances in targeted therapies are further improving treatment protocols. The next-generation anti-HER2 monoclonal antibody margetuximab targets the same HER2 epitope as trastuzumab and is modified in its Fc domain to improve antibody-dependent cellular cytotoxicity.[6] Margetuximab was approved by the FDA in December 2020 for the treatment of metastatic breast cancer in patients who had received prior HER2 therapies[7]. Two different bispecific antibodies have also been developed and are in clinical trials: ZW25 is a bispecific antibody that targets two different HER2 epitopes, and PRS-343 is a bispecific antibody that targets a HER2 epitope and the 4-1BB costimulatory immunoreceptor[8]. Bispecific antibodies are under development for breast cancer and other cancers because they exploit the property that antibodies have two epitope binding sites and can be engineered to better target a single molecule or crosslink multiple molecules, like HER2 and immune checkpoint molecules. Antibody-drug conjugates have also been evaluated as novel therapeutics that target HER2. Trastuzumab deruxtecan is an anti-HER2 antibody conjugated to a cytotoxic payload that is linked by a cleavable drug linker[9]. This treatment has been recently approved by the FDA and it shows superior targeting of cytotoxic drugs to HER2+ tumor cells and shows efficacy on individuals who have been treated previously with anti-HER2 mAbs. HER2-specific tyrosine kinase inhibitors have also been successful in treating HER2+ tumors, especially brain metastases because these drugs can cross the blood-brain barrier. Lapatinib, neratinib, pyrotinib, and tucatinib are four HER2-specific tyrosine kinase inhibitors used to treat HER2+ breast cancers. Lapatinib is an oral, irreversible small molecule HER1/HER2/HER4 TKI that was first approved in 2007 for the treatment of metastatic breast cancer[10]. Neratinib is a similar, orally available irreversible small molecule HER1/HER2/HER4 TKI that showed better efficacy against EGFR mutants and has been approved for use as an adjuvant therapy following trastuzumab treatment[11]. The development of resistance is also a problem for HER2-targeted TKIs, and pyrotinib is an experimental HER1/HER2/HER4 TKI under investigation as a potential new drug that is less prone to resistance[12]. Tucatinib is a TKI that is highly selective for HER2 approved for use in combination with trastuzumab and capecitabine in HER2+ breast and colorectal cancers, including patients with metastases and who have received previous anti-HER2 treatment[13]. HER2+ breast cancers continue to be challenging to treat due to resistance and metastasis. Combination therapies with antibodies and targeted inhibitors have dramatically improved survival in HER2+ breast cancer patients, but individuals who relapse continue to be faced with limited treatment options. New antibody-drug conjugates, bispecific antibodies, and targeted inhibitors are under investigation and hold the promise of better, more durable treatment options for patients. [1] Hudis CA. Trastuzumab--mechanism of action and use in clinical practice. N. Engl. J. Med. 2007 Jul 5;357(1):39-51. [2] Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987 Jan 9;235(4785):177-82. [3] Moasser MM. The oncogene HER2: its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene. 2007 Oct 4;26(45):6469-87. [4] Romond EH, Perez EA, Bryant J, Suman VJ, Geyer Jr CE, Davidson NE, Tan-Chiu E, Martino S, Paik S, Kaufman PA, Swain SM. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N. Engl. J. Med. 2005 Oct 20;353(16):1673-84. [5] Smith I, Procter M, Gelber RD, Guillaume S, Feyereislova A, Dowsett M, Goldhirsch A, Untch M, Mariani G, Baselga J, Kaufmann M. 2-year follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer: a randomised controlled trial. Lancet. 2007 Jan 6;369(9555):29-36. [6] Rugo HS, Im SA, Cardoso F, Cortés J, Curigliano G, Musolino A, Pegram MD, Wright GS, Saura C, Escrivá-de-Romaní S, De Laurentiis M. Efficacy of margetuximab vs trastuzumab in patients with pretreated ERBB2-positive advanced breast cancer: a phase 3 randomized clinical trial. JAMA Oncol. 2021 Apr 1;7(4):573-84 [7] https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-margetuximab-metastatic-her2-positive-breast-cancer. [8] Kunte S, Abraham J, Montero AJ. Novel HER2-targeted therapies for HER2-positive metastatic breast cancer. Cancer. 2020 Oct 1;126(19):4278-4288. [9] Doi T, Shitara K, Naito Y, Shimomura A, Fujiwara Y, Yonemori K, Shimizu C, Shimoi T, Kuboki Y, Matsubara N, Kitano A, Jikoh T, Lee C, Fujisaki Y, Ogitani Y, Yver A, Tamura K. Safety, pharmacokinetics, and antitumour activity of trastuzumab deruxtecan (DS-8201), a HER2-targeting antibody-drug conjugate, in patients with advanced breast and gastric or gastro-oesophageal tumours: a phase 1 dose-escalation study. Lancet Oncol. 2017 Nov;18(11):1512-1522. [10] Ryan Q, Ibrahim A, Cohen MH, Johnson J, Ko CW, Sridhara R, Justice R, Pazdur R. FDA drug approval summary: lapatinib in combination with capecitabine for previously treated metastatic breast cancer that overexpresses HER-2. Oncologist. 2008 Oct;13(10):1114-9. [11] Minami Y, Shimamura T, Shah K, LaFramboise T, Glatt KA, Liniker E, Borgman CL, Haringsma HJ, Feng W, Weir BA, Lowell AM, Lee JC, Wolf J, Shapiro GI, Wong KK, Meyerson M, Thomas RK. The major lung cancer-derived mutants of ERBB2 are oncogenic and are associated with sensitivity to the irreversible EGFR/ERBB2 inhibitor HKI-272. Oncogene. 2007 Jul 26;26(34):5023-7. [12] Li Q, Guan X, Chen S, Yi Z, Lan B, Xing P, Fan Y, Wang J, Luo Y, Yuan P, Cai R, Zhang P, Li Q, Zhong D, Zhang Y, Zou J, Zhu X, Ma F, Xu B. Safety, Efficacy, and Biomarker Analysis of Pyrotinib in Combination with Capecitabine in HER2-Positive Metastatic Breast Cancer Patients: A Phase I Clinical Trial. Clin. Cancer Res. 2019 Sep 1;25(17):5212-5220. [13] Murthy RK, Loi S, Okines A, Paplomata E, Hamilton E, Hurvitz SA, Lin NU, Borges V, Abramson V, Anders C, Bedard PL, Oliveira M, Jakobsen E, Bachelot T, Shachar SS, Müller V, Braga S, Duhoux FP, Greil R, Cameron D, Carey LA, Curigliano G, Gelmon K, Hortobagyi G, Krop I, Loibl S, Pegram M, Slamon D, Palanca-Wessels MC, Walker L, Feng W, Winer EP. Tucatinib, Trastuzumab, and Capecitabine for HER2-Positive Metastatic Breast Cancer. N Engl J Med. 2020 Feb 13;382(7):597-609.
Prostate Cancer - Drivers of Genetic Mutations & Their Role in Disease Progression
Prostate cancer is one of the most common forms of cancer that affects the prostate gland in the male urogenital tract. Most prostate cancers are slow growing and are usually detected in individuals older than age 50. Although environmental factors contribute to the development of prostate cancer, risk is significantly associated with incidence among first-degree relatives. In recent years, several inherited and spontaneously mutated genes have been linked to prostate cancer. Here we highlight these findings and how they provide insight into the development of targeted prostate cancer therapies. Prostate cancer tumors are typically characterized by a wide range of genomic aberrations including somatic copy number alterations (SCNAs), structural rearrangements, and point mutations. Metastatic prostate tumors may have hundreds of these aberrations throughout the genome, but recent studies of primary prostate tumors have been critical to defining mutations associated with tumor development. Most primary prostate tumors have SCNAs, typically in the form of deletions that target a small area of the genome1. Deletions are often seen in the tumor suppressor genes BRCA2, NKX3.1, PTEN, and RB12. Structural rearrangements are another source of genomic variation observed in primary prostate tumors, for which the TMPRSS2:ERG rearrangement is most common and causes the androgen-responsive TMPRSS2 serine protease to drive expression of the ERG oncogene3. Other rearrangements have been linked to prostate cancer, including TMPRSS2 fusions with other ETS family members beyond ERG (ETV1 and ETV4)4. Some studies have suggested a link between ERG rearrangements and SCNA incidence, although it is not yet clear if rearrangements lead to destabilization of the genome and increased occurrence of SCNAs1. Somatic mutations occur on relatively high frequency primary prostate tumors, and this has been linked to mutations DNA mismatch repair proteins, tumor suppressors and oncogenes. The tumor suppressor TP53 is consistently identified as mutated in prostate tumors5. Mutations in the tumor suppressor genes BRCA1 and BRCA2 are well known for their association with breast and ovarian cancer but have also been linked to prostate cancer, and recent studies have shown that certain BRCA2 pathogenic sequence variants are associated with an elevated risk for aggressive prostate cancer6,7. PALB2 is a protein that interacts with BRCA2, and a recent study indicated that individuals who have certain PALB2 variants are more predisposed to aggressive forms of prostate cancer8. In addition, mutations in genes associated with DNA mismatch repair (MMR), such as MSH2, MSH6, and MLH1, are associated with greater prostate cancer risk9, and individuals with Lynch syndrome, which is caused by a germline mutation in one of these MMR genes, are also at greater risk of developing an aggressive form of prostate cancer10. One of the most important genes involved in prostate cancer progress is the gene expressing the androgen receptor (AR). Dozens of somatic mutations have been identified in the AR, and many of these mutations target the ligand binding domain11. Some prostate cancers can be treated successfully with drugs that inhibit AR signaling, such as enzalutamide, but resistance frequently occurs and is associated with worse outcomes. AR promotes prostate cancer cell survival by regulating several cellular programs. Although AR activity inhibition through androgen deprivation therapy (ADT) results in the suppression of AR-related pathways and durable clinical remission12, the disease may recur as castration-resistant prostate cancer (CRPC), typically with reactivated AR signaling. In this case, patients are treated with second-generation AR pathway inhibitors such as enzalutamide and abiraterone. Unfortunately, durable complete responses under these treatments are rare, and a substantial number of CRPC tumors progress under treatment despite loss of AR signaling. CRPC with AR-null phenotype are classified as tumors with diffuse small cell or neuroendocrine (NE) characteristics (SCNPC) or as double-negative (DNPC) phenotype that lacks both NE and AR activity13. A recently described genome-wide CRISPR-Cas9 screen was used to identify kinases that could be inhibited in the presence of enzalutamide. BRAF or downstream MAPK signaling molecules were identified as targets that could be co-inhibited with enzalutamide to improve overall therapeutic effects of these drugs14. Genomic alterations in prostate cancer are highly variable and can impact many critical cellular processes. By understanding which genes drive disease progression, potential therapeutic targets can be identified, and disease outcomes under different treatment regimens can be better predicted. [1] Taylor BS, Schultz N, Hieronymus H, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010 Jul 13;18(1):11-22. [2] Wallis CJ, Nam RK. Prostate Cancer Genetics: A Review. EJIFCC. 2015 Mar 10;26(2):79-91. [3] Tomlins SA, Rhodes DR, Perner S et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005 Oct 28;310(5748):644-8. [4] Rubin MA, Maher CA, Chinnaiyan AM. Common gene rearrangements in prostate cancer. J. Clin. Oncol. 2011 Sep 20;29(27):3659-68. [5] Ecke TH, Schlechte HH, Schiemenz K et al. TP53 gene mutations in prostate cancer progression. Anticancer Res. 2010 May 1;30(5):1579-86. [6] Patel VL, Busch EL, Friebel TM, et al. Association of Genomic Domains in BRCA1 and BRCA2 with Prostate Cancer Risk and Aggressiveness. Cancer Res. 2020 Feb 1;80(3):624-638. [7] Oh M, Alkhushaym N, Fallatah S, et al. The association of BRCA1 and BRCA2 mutations with prostate cancer risk, frequency, and mortality: A meta-analysis. Prostate. 2019 Jun;79(8):880-895. [8] Wokołorczyk D, Kluźniak W, Stempa K, Rusak B, Huzarski T, Gronwald J, Gliniewicz K, Kashyap A, Morawska S, Dębniak T, Jakubowska A. PALB2 mutations and prostate cancer risk and survival. Br. J. Cancer. 2021 May 18:1-7. [9] Ritch E, Fu SY, Herberts C, Wang G, et al. Identification of hypermutation and defective mismatch repair in ctDNA from metastatic prostate cancer. Clin. Cancer Res. 2020 Mar 1;26(5):1114-25. [10] Haraldsdottir S, Hampel H, Wei L, et al. Prostate cancer incidence in males with Lynch syndrome. Genet. Med. 2014 Jul;16(7):553-7. [11] Ferraldeschi R, Welti J, Luo J, Attard G, de Bono JS. Targeting the androgen receptor pathway in castration-resistant prostate cancer: progresses and prospects. Oncogene. 2015 Apr 2;34(14):1745-57. [12] Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocr Rev. 2004;25(2):276–308. [13] Bluemn EG, Coleman IM, Lucas JM et al. Androgen receptor pathway-independent prostate cancer is sustained through FGF signaling. Cancer Cell. 2017;32(4):474–489. [14] Palit SAL, van Dorp J, Vis D, Lieftink C, Linder S, Beijersbergen R, Bergman AM, Zwart W, van der Heijden MS. A kinome-centered CRISPR-Cas9 screen identifies activated BRAF to modulate enzalutamide resistance with potential therapeutic implications in BRAF-mutated prostate cancer. Sci. Rep. 2021 Jul 1;11(1):13683. Subscribe to our Trends in Oncology Blog
The Benefits of Cell Line-Derived Xenograft Models for Preclinical Oncology Screening
Preclinical oncology research is a critical component in the development of new cancer therapeutics, and it is essential that drug candidates are carefully screened before they are tested in human clinical trials. One valuable tool in preclinical oncology research is the use of cell line-derived xenograft (CDX) models. In this blog post, we will explore the benefits of CDX models for preclinical oncology screening and how they can be used to improve our understanding of cancer and the development of new treatments. CDX models are created by implanting human cancer cells into immunocompromised mice. These cells develop into tumors that resemble their human counterparts and can be used to study the biology of cancer and test new treatments. One significant advantage of CDX models is that they are relatively easy and fast to establish, making them a cost-effective alternative to other preclinical screening models.[1] CDX models allow researchers to study the response of tumors to treatments in vivo, replicating the conditions of human cancer. Additionally, they can be used to evaluate the pharmacodynamics and pharmacokinetics (PD/PK) of candidate drugs. These models allow researchers to evaluate how drugs impact tumor growth over time, and how the drug is distributed throughout the body. This information is critical in determining the optimal dosing regimen for cancer patients.[2-4] CDX models have been widely used in preclinical research for several decades and can provide valuable insights into the underlying biology of cancer. Tumors grown in CDX models can be analyzed in depth to identify biological markers that can inform the development of targeted therapies. The use of CDX models can help researchers better understand the complexity of cancer and identify new therapeutic targets.[5-6] In conclusion, CDX models offer several advantages for preclinical oncology screening, including their ease of establishment, ability to replicate human conditions, and ability to provide insights into the biology of cancer. By incorporating CDX models into their preclinical screening strategies, researchers can more accurately predict the efficacy of cancer drugs and improve the development of new cancer therapies. 1. Long Y, Xie B, Shen HC, Wen D. Translation Potential and Challenges of In Vitro and Murine Models in Cancer Clinic. Cells. 2022 Nov 30;11(23):3868. doi: 10.3390/cells11233868. PMID: 36497126; PMCID: PMC9741314. 2. Qin L, Wang L, Zhang J, Zhou H, Yang Z, Wang Y, Cai W, Wen F, Jiang X, Zhang T, Ye H, Long B, Qin J, Shi W, Guan X, Yu Z, Yang J, Wang Q, Jiao Z. Therapeutic strategies targeting uPAR potentiate anti-PD-1 efficacy in diffuse-type gastric cancer. Sci Adv. 2022 May 27;8(21):eabn3774. doi: 10.1126/sciadv.abn3774. Epub 2022 May 25. PMID: 35613265; PMCID: PMC9132454. 3. Chen Y, Chen HN, Wang K, Zhang L, Huang Z, Liu J, Zhang Z, Luo M, Lei Y, Peng Y, Zhou ZG, Wei Y, Huang C. Ketoconazole exacerbates mitophagy to induce apoptosis by downregulating cyclooxygenase-2 in hepatocellular carcinoma. J Hepatol. 2019 Jan;70(1):66-77. doi: 10.1016/j.jhep.2018.09.022. Epub 2018 Oct 1. PMID: 30287340. 4. Kendsersky NM, Lindsay J, Kolb EA, Smith MA, Teicher BA, Erickson SW, Earley EJ, Mosse YP, Martinez D, Pogoriler J, Krytska K, Patel K, Groff D, Tsang M, Ghilu S, Wang Y, Seaman S, Feng Y, Croix BS, Gorlick R, Kurmasheva R, Houghton PJ, Maris JM. The B7-H3-Targeting Antibody-Drug Conjugate m276-SL-PBD Is Potently Effective Against Pediatric Cancer Preclinical Solid Tumor Models. Clin Cancer Res. 2021 May 15;27(10):2938-2946. doi: 10.1158/1078-0432.CCR-20-4221. Epub 2021 Feb 22. PMID: 33619171; PMCID: PMC8127361. 5. Oduwole OO, Poliandri A, Okolo A, Rawson P, Doroszko M, Chrusciel M, Rahman NA, Serrano de Almeida G, Bevan CL, Koechling W, Huhtaniemi IT. Follicle-stimulating hormone promotes growth of human prostate cancer cell line-derived tumor xenografts. FASEB J. 2021 Apr;35(4):e21464. doi: 10.1096/fj.202002168RR. PMID: 33724574. 6. Tang N, Cheng C, Zhang X, Qiao M, Li N, Mu W, Wei XF, Han W, Wang H. TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight. 2020 Feb 27;5(4):e133977. doi: 10.1172/jci.insight.133977. PMID: 31999649; PMCID: PMC7101140.
ERK at Work - Targeting the ERK Pathway in Cancer
Signal transduction networks are essential to normal cell growth and function, but signaling abnormalities, such as those caused by spontaneous mutations in signaling components, are the leading causes of tumor growth. The extracellular signal-regulated kinase (ERK) signaling pathway contributes to normal cell proliferation, differentiation, and survival, but components of the ERK pathway are upregulated in many tumor types, including those associated with melanoma, lung, colon and pancreatic cancers. The critical role of the ERK pathway in cancer has resulted in the development of drugs and therapeutics that can inhibit overactivation of ERK signaling molecules or target aberrant signaling pathways upstream of ERK. The emergence of precision medicine-driven cancer treatments has advanced the ERK pathway inhibitor field. Breakthroughs in cancer genomics has enabled researchers to analyze tumor sequences derived from patient data and identify novel targets in the ERK pathway. This type of computational analysis coupled with large scale in vitro screening is essential to narrowing down which novel inhibitors are studied further during preclinical development. The use of patient-derived xenograft (PDX) mouse models is critical to the discovery of next-generation ERK inhibitors. PDX models involve the use of mice that are engrafted with tumor tissue derived from patient samples. These mice typically have suppressed immune systems in order to tolerate tumor grafts and can be engineered to express other components of the human immune system, such as immune checkpoint inhibitors. PDX models that use tumors with mutations in ERK signaling, such as a colorectal tumor with a V600-mutated BRAF molecule, can be used in the preclinical screening of novel ERK pathway inhibitors. Novel inhibitors can be screened simultaneously in different PDX models, and the safety and efficacy of these molecules can also be assessed in combination with other therapeutics. We are entering an era when cancer patients will have access to precision medicine treatments informed by the genetic analysis of their tumors, and the strategic deployment of new and improved ERK pathway inhibitors in patients with specific ERK mutations will improve treatment efficacy and overall survival. PDX models are essential to the development of these new ERK pathway inhibitors and many other novel immunotherapies.

Unveiling the Secrets of PBMC Isolation Tubes: Clinching Clinical Trial Success with the Right Tools
The quality of data received from clinical specialty testing labs is largely dependent on the integrity of the samples tested. One critical aspect of this is the proper isolation of PBMCs - peripheral blood mononuclear cells - often used in high-complexity flow cytometry research. To get the best quality results, researchers must use the most optimal tubes to isolate these cells. Three main types of tubes are commonly used: SepMate tubes, CPT tubes, and Manual Ficoll Gradient tubes. Each of these tubes has advantages and disadvantages that researchers must consider carefully. In this blog post, we will look at the pros and cons of each tube type and what you should consider before selecting one for your clinical trial. SepMate Tubes: SepMate tubes come in different sizes that enable researchers to isolate PBMCs from different numbers of tubes at once. These tubes work by using an inserted plastic separation technology that helps remove unwanted clumps of cells. One of the most significant advantages of these tubes is that they can be used for small to medium-sized samples with ease. The technology also separates cells significantly, making it easy to measure the concentration of cells after the separation procedure accurately. Furthermore, the tube also allows for rapid and consistent layering and easy collection of PBMCs, making it ideal for research. However, obtaining a high percentage of neutrophils is not possible, as they would be trapped below the barrier inside the tube. SepMate tubes are disposable, which makes them less financially sensible in the long run. CPT Tubes: CPT tubes are anticoagulant-based layered tubes that allow for the collection directly to cell separation without any additional processing. CPT tubes simplify the separation process and reduce the risk of contamination. They offer a more robust PBMC isolation process, and specific manufacturers produce four types to suit different research needs regarding testing human cells. The tubes generate high PBMC yields with high cell viability, making them an optimal option where quality is paramount. The primary disadvantage of CPT tubes is that they are quite expensive when compared to other tube types for PBMC isolation. Manual Ficoll Gradient Tubes: Manual Ficoll Gradient tubes require manual handling and processing, so they aren't automated. Due to this, the individual processing of Ficoll gradient tubes can cause variability in the manual layering steps, leading to variation in the final cell yield. The biggest issue with this type of isolation tube is that it takes longer to obtain the actual PBMCs than using SepMate or CPT tubes, which may create more significant delays in your research. Additionally, due to the manual nature of this type of PBMC isolation, final PBMC recovery rates are usually dependent on the experience of the researcher. In conclusion, the right tube type will depend on your primary requirements for your research. Regardless of your choice, there is still a need to make sure you select a tube type that meets the technical requirements of your studies and delivers high-quality results. Champions Oncology has expertise in GCLP-compliant PBMC isolation using SepMate, CPT, and manual Ficoll Gradient tube types and can advise the best choice for your upcoming clinical trial.
Sarcoma Immunotherapeutic Strategies
Sarcomas are a group of aggressive heterogeneous tumors for which more than 100 histological subtypes have been defined1. Sarcomas are found in a variety of solid tissues, including bone and gastrointestinal stromal cells. Current treatment options include radiotherapy, surgical resection, targeted therapies, and chemotherapy, but these treatments have had limited efficacy on intermediate to high-grade tumors. Investigations into the molecular and cellular mechanisms that drive sarcomas have helped identify potential biomarkers that can serve as potential therapeutic targets. In addition, recent studies have also focused on the tumor microenvironment (TME) within sarcomas and the roles of different immune cell subsets creating an immunosuppressive microenvironment. These observations directly inform novel immunotherapeutic approaches that are being examined in preclinical and clinical studies. Sarcoma TME and Immune Checkpoint Blockade In general, sarcoma TMEs are highly immunosuppressive environments, and a recent analysis of the Ewing’s sarcoma family of tumors (ESFT) showed poor overall survival correlated with tumors that had a TME with elevated expression of hypoxia-inducible factor 1-α and were enriched with immunosuppressive M2 macrophages and neutrophils2. The presence of tumor-associated macrophages has also been linked to sarcoma growth and metastasis, and elevated expression of the immune checkpoint molecule PD-L1 also correlated with metastasis and poor overall survival3,4. These observations led to clinical trials that evaluated the efficacy of immune checkpoint blockade for treating sarcomas, but treatments that targeted PD-1/PD-L1 or CTLA-4 showed limited clinical efficacy and did not meet primary endpoints for overall survival5,6. Targeted Therapies Genomic studies of sarcomas have been critical to identifying mutations associated with tumor growth, angiogenesis, and metastasis. These findings led to clinical trials examining the efficacy of tyrosine kinase inhibitors or inhibitors that target vascular endothelial growth factor or insulin-like growth factor-1, but most of these trials failed to meet overall survival endpoints7,8. More recent genomic studies have indicated that mutations associated with different tumors occur in the same genes as some of the targeted therapies, but the mutations differ sufficiently to render these therapies ineffective9. Cellular Immunotherapy Cell-based immunotherapies have shown great promise in treating hematologic malignancies and are being explored for solid tumors. Chimeric antigen receptor (CAR) T cells have been of particular interest as they are patient-derived T cells engineered to express a tumor-specific receptor and thus can target tumors without being detected as foreign cells. Preclinical studies suggested that sarcoma-specific CAR T cells may be effective, but clinical trials have failed due to poor penetration and function of CAR T cells in the immunosuppressive TME10. Recent and ongoing studies are exploring the effects of multi-CAR T cell strategies that target different tumor antigens, as well as the effects of using modified dendritic cells or natural killer cells or combining treatment modalities11. Effective and lasting treatment options for sarcomas continue to be elusive but advances in genomic analysis and immune-based treatments are slowly improving outcomes. Like other cancers, clinical studies exploring treatment combinations are also showing promise and may provide critical breakthroughs for some of the most aggressive sarcomas. 1 Doyle LA. Sarcoma classification: an update based on the 2013 World Health Organization Classification of Tumors of Soft Tissue and Bone. Cancer. 2014 Jun 15;120(12):1763-74. 2 Stahl D, Gentles AJ, Thiele R, Gütgemann I. Prognostic profiling of the immune cell microenvironment in Ewing’s Sarcoma Family of Tumors. Oncoimmunology. 2019 Dec 2;8(12):e1674113. 3 Xiao W, Klement JD, Lu C, Ibrahim ML, Liu K. IFNAR1 controls autocrine type I IFN regulation of PD-L1 expression in myeloid-derived suppressor cells. J. Immunol. 2018 Jul 1;201(1):264-77. 4 Zhu Z, Jin Z, Zhang M, Tang Y, Yang G, Yuan X, Yao J, Sun D. Prognostic value of programmed death-ligand 1 in sarcoma: a meta-analysis. Oncotarget. 2017 Aug 29;8(35):59570. 5 Maki RG, Jungbluth AA, Gnjatic S, Schwartz GK, D’Adamo DR, Keohan ML, Wagner MJ, Scheu K, Chiu R, Ritter E, Kachel J. A pilot study of anti-CTLA4 antibody ipilimumab in patients with synovial sarcoma. Sarcoma. 2013 Oct;2013. 6 Zuo W and Lingdi Z. Recent advances and application of PD-1 blockade in sarcoma." Oncotargets and Therapy 2019; 12:6887. 7 Ray-Coquard IL, Domont J, Tresch-Bruneel E, Bompas E, Cassier PA, Mir O, Piperno-Neumann S, Italiano A, Chevreau C, Cupissol D, Bertucci F. Paclitaxel given once per week with or without bevacizumab in patients with advanced angiosarcoma: a randomized phase II trial. J. Clin. Oncol. 2015 Sep 1;33(25):2797-802. 8 Tap WD, Demetri G, Barnette P, Desai J, Kavan P, Tozer R, Benedetto PW, Friberg G, Deng H, McCaffery I, Leitch I. Phase II study of Ganitumab, a fully human anti–type-1 insulin-like growth factor receptor antibody, in patients with metastatic ewing family tumors or desmoplastic small round cell tumors. J. Clin. Oncol. 2012 May 20;30(15):1849-56. 9 Trédan O, Wang Q, Pissaloux D, Cassier P, de la Fouchardière A, Fayette J, Desseigne F, Ray-Coquard I, de la Fouchardière C, Frappaz D, Heudel PE. Molecular screening program to select molecular-based recommended therapies for metastatic cancer patients: analysis from the ProfiLER trial. Ann. Oncol. 2019 May 1;30(5):757-65. 10 Thanindratarn P, Dean DC, Nelson SD, Hornicek FJ, Duan Z. Chimeric antigen receptor T (CAR-T) cell immunotherapy for sarcomas: From mechanisms to potential clinical applications. Cancer Treatment Reviews. 2020 Jan 1; 82:101934. 11 Patel S, Burga RA, Powell AB, Chorvinsky EA, Hoq N, McCormack SE, Van Pelt SN, Hanley PJ, Cruz CR. Beyond CAR T cells: other cell-based immunotherapeutic strategies against cancer. Front. Onc. 2019 Apr 10; 9:196.
PARP Inhibitors for Ovarian Cancer Treatment
Ovarian cancer remains one of the leading causes of mortality for women with gynecological cancers and continues to be associated with stubbornly low 5-year survival rates. Depending on the stage or type of ovarian cancer, localized treatments including surgical resection or radiation may be sufficient. Treatment of metastatic ovarian cancer may include surgery and radiation combined with chemotherapy. Hormone therapy is also typically combined with chemotherapy for targeted reduction of estrogen and/or stimulation of progesterone for the targeted treatment of ovarian stromal tumors that produce high levels of estrogen. Nonetheless, many of these treatments have limited efficacy for treating metastatic ovarian cancer. Advances in targeted ovarian cancer treatments have been facilitated by identification of cellular replication machinery in some cancer subtypes, including defects in the breast cancer-associated protein (BRCA)1 and BRCA2 genes, which play critical roles in DNA repair and transcription regulation[1]. Poly (ADP-ribose) polymerases (PARP) are a family of enzymes that can repair single strand DNA breaks through the base excision repair pathway[2]. PARPs can work together with normal BRCA1/2 to repair double strand DNA breaks by the homologous recombination (HR) pathway. Preclinical studies have shown that PARP inhibitors are effective in cells with BRCA1/2 mutations because these mutations combined with PARP inhibition cause synthetic lethality due to blockade of multiple DNA repair pathways. Olaparib was the first PARP inhibitor approved for the treatment of germline BRCA1/2-mutated metastatic ovarian cancer in 2014[3]. This treatment was recommended for patients who had undergone three or more rounds of chemotherapy. Olaparib was later approved as a first-line maintenance therapy for germline or somatic BRCA1/2 mutations as well for the treatment of advanced ovarian cancers that have partial or complete responses to first-line platinum-based chemotherapies. Two other PARP inhibitors are also currently approved for different ovarian cancer diagnoses including rucaparib, which is approved for the treatment of germline BRCA1/2 mutations following two rounds of chemotherapy and for maintenance therapy following platinum-based therapies regardless of BRCA mutation status. Similarly, niraparib is approved to treat ovarian cancers with BRCA1/2 mutations or genomic instability and disease progression following platinum-based chemotherapies. Like many other targeted therapies, some patients are completely unresponsive to treatment or develop resistance to PARP inhibitor monotherapy. Recent studies have described promising results associated with combined treatments, including PARP inhibitors combined with anti-angiogenic drugs like bevacizumab[4] or immune check point inhibitors like pembrolizumab[5]. There are currently three ongoing phase III first-line trials comparing the efficacy of combining PARP inhibitors and immune checkpoint blockade with VEGF inhibitors. Several thousand women are being enrolled in these studies with a goal of trying to determine if these combinations improve outcomes[6]. PARP inhibitors continue to be effective for treating hormone receptor-negative tumors but are less effective in treating hormone-receptor-positive ovarian tumors. Current trials are exploring the use of hormone inhibitors to disrupt hormone receptor signaling in ovarian tumors and render them sensitive to PARP inhibition[7]. PARP inhibitors have provided clinical benefits to ovarian cancer patients who previously had limited treatment options. Future studies will likely improve the efficacy of PARP inhibition as a monotherapy or in combination with other targeted therapies. [1] Venkitaraman AR. Functions of BRCA1 and BRCA2 in the biological response to DNA damage. J Cell Sci. 2001 Oct;114(Pt 20):3591-8. [2] D'Amours D, Desnoyers S, D'Silva I, Poirier GG. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J. 1999 Sep 1;342 ( Pt 2)(Pt 2):249-68. [3] Franzese E, Centonze S, Diana A, Carlino F, Guerrera LP, Di Napoli M, De Vita F, Pignata S, Ciardiello F, Orditura M. PARP inhibitors in ovarian cancer. Cancer Treatment Reviews. 2019 Feb 1;73:1-9. [4] Ray-Coquard I, Pautier P, Pignata S, Pérol D, González-Martín A, Berger R, Fujiwara K, Vergote I, Colombo N, Mäenpää J, Selle F. Olaparib plus bevacizumab as first-line maintenance in ovarian cancer. New England Journal of Medicine. 2019 Dec 19;381(25):2416-28. [5] Konstantinopoulos PA, Waggoner S, Vidal GA, Mita M, Moroney JW, Holloway R, Van Le L, Sachdev JC, Chapman-Davis E, Colon-Otero G, Penson RT, Matulonis UA, Kim YB, Moore KN, Swisher EM, Färkkilä A, D'Andrea A, Stringer-Reasor E, Wang J, Buerstatte N, Arora S, Graham JR, Bobilev D, Dezube BJ, Munster P. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination with Pembrolizumab in Patients With Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol. 2019 Aug 1;5(8):1141-1149. [6] Elyashiv, Osnat, Yien Ning Sophia Wong, and Jonathan A. Ledermann. "Frontline Maintenance Treatment for Ovarian Cancer." Current Oncology Reports 23.8 (2021): 1-10.m [7] Matulonis UA, Wulf GM, Barry WT, Birrer M, Westin SN, Farooq S, Bell-McGuinn KM, Obermayer E, Whalen C, Spagnoletti T, Luo W. Phase I dose escalation study of the PI3kinase pathway inhibitor BKM120 and the oral poly (ADP ribose) polymerase (PARP) inhibitor olaparib for the treatment of high-grade serous ovarian and breast cancer. Annals of Oncology. 2017 Mar 1;28(3):512-8.

RNAseq and Whole Transcriptome Sequencing: Understanding Gene Expression Analysis
With advances in next-generation sequencing (NGS) technologies, gene expression analysis has become an increasingly popular area of research. Two common methods used for this purpose are RNA sequencing (RNAseq) and whole transcriptome sequencing (WTS). Both techniques provide valuable insights into the molecular mechanisms of individual cells and organisms. However, when should one use RNAseq versus WTS for gene expression analysis? In this post, we will explore the advantages and disadvantages of both methods and provide guidance on which one to use. RNAseq RNAseq is a method of sequencing RNA molecules and measuring the abundance of each transcript. This method is capable of identifying all types of RNA, including messenger RNA (mRNA), noncoding RNA, and small RNA. It requires high-quality RNA and can detect alternative splicing, fusion genes, and post-transcriptional modifications. RNAseq is particularly useful for identifying novel transcripts and quantifying gene expression levels in low-abundance genes[1]. However, RNAseq has some disadvantages. Its high sensitivity may lead to high levels of noise when dealing with lowly expressed genes. The method can also be expensive, and the data analysis process can be complex. Additional bioinformatic tools are required to accurately quantify gene expression levels, identify differential expression, and perform pathway analysis[1]. Whole Transcriptome Sequencing WTS is a newer approach that can comprehensively analyze the transcriptome of an organism without relying on annotation. This method sequences the entire complement of RNA sequences, including both known and unknown transcripts. WTS can provide greater resolution for splice variants and more accurate gene expression quantification compared to RNAseq[2]. WTS has several advantages, including the elimination of the need for gene annotation. The technique is also cost-effective since the sequencing depth can be adjusted based on the number of samples analyzed. The method is flexible and can be used in a variety of applications such as identifying regulatory noncoding RNAs and studying alternative splicing events[2]. However, WTS comes with a few drawbacks. The technique requires higher sequencing depth for accurate gene expression quantification, which can increase the cost of the experiment. Additionally, WTS can detect unexpected transcripts or splice variants, creating new challenges for data analysis and interpretation[2]. When to use RNAseq versus WTS Choosing between RNAseq and WTS depends on the goals of the experiment, the type of RNA to be analyzed, and the available resources. Researchers should consider the complexity of their sample and the genotypic variation, as WTS is better suited for samples with a higher degree of variability. RNAseq is a better choice for studies involving differential expression of known protein-coding genes. In contrast, WTS can identify novel transcripts and regulatory noncoding RNA without relying on a predetermined set of annotated transcripts[3-4]. In conclusion, RNAseq and WTS are both powerful techniques for gene expression analysis. RNAseq is better suited for identifying differentially expressed genes in complex samples with low-abundance genes, while WTS is more effective when searching for novel transcripts and studying alternative splicing events. Both techniques require careful experimental design and bioinformatic analysis for accurate results. Ultimately, researchers should choose the method that aligns with their experimental goals and available resources. Understanding the advantages and disadvantages of each method can help optimize experimental design and result interpretation. At Champions Oncology, we offer both RNAseq and WTS. We use an enrichment-based RNAseq method when the input RNA is of high quality, while we opt for a depletion-based WTS method when the input RNA quality is lower, with an integrity score as low as 2.0. This approach allows us to utilize different methods in different situations to obtain gene expression results from a broader range of samples. [1] Martin JA, Wang Z. Next-generation transcriptome assembly. Nat Rev Genet. 2011 Sep 7;12(10):671-82. doi: 10.1038/nrg3068. PMID: 21897427. [2] Jobanputra V, Wrzeszczynski KO, Buttner R, Caldas C, Cuppen E, Grimmond S, Haferlach T, Mullighan C, Schuh A, Elemento O. Clinical interpretation of whole-genome and whole-transcriptome sequencing for precision oncology. Semin Cancer Biol. 2022 Sep;84:23-31. doi: 10.1016/j.semcancer.2021.07.003. Epub 2021 Jul 10. PMID: 34256129. [3] Fu X, Fu N, Guo S, Yan Z, Xu Y, Hu H, Menzel C, Chen W, Li Y, Zeng R, Khaitovich P. Estimating accuracy of RNA-Seq and microarrays with proteomics. BMC Genomics. 2009 Apr 16;10:161. doi: 10.1186/1471-2164-10-161. PMID: 19371429; PMCID: PMC2676304. [4] Ruan M, Liu J, Ren X, Li C, Zhao AZ, Li L, Yang H, Dai Y, Wang Y. Whole transcriptome sequencing analyses of DHA treated glioblastoma cells. J Neurol Sci. 2019 Jan 15;396:247-253. doi: 10.1016/j.jns.2018.11.027. Epub 2018 Nov 22. PMID: 30529802.

Current Trends in Gastrointestinal Cancer – CRISPR/Cas9-based Immunotherapy
Gastrointestinal cancers are a heterogeneous group of cancers that can be caused by H. pylori bacterial or Epstein-Barr virus infection, chronic inflammation, or inherited or spontaneous genetic mutations. Advances in solid tumor immunotherapy for gastrointestinal cancers include the development of methods that improve the screening, evaluation, and development of more precise and robust treatments. Breakthroughs in basic and translational research are leading to better treatment options for gastrointestinal cancer, and CRISPR/Cas9-based (clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9) gene editing technology is at the forefront of these discoveries. CRISPR/Cas9-based gene editing technology was originally identified in bacteria and archaea as an antiviral defense mechanism[1]. As a gene editing application, CRISPR/Cas9 uses the Cas9 nuclease to cleave specific sequences in the target DNA and introduces modifications such as knock-in or knock-out mutations[2]. The DNA sequence is targeted using a single-guide RNA (sgRNA) comprised of CRISPR RNA (crRNA) and trans-activating CRISPR RNA (tracRNA), and these RNAs can be expressed on the same plasmid as the Cas9 nuclease upon delivery into target cells. The crRNA/tracrRNA duplex can bind to the complementary sequence in the genomic DNA and recruit Cas9, which cleaves the target sequence and allows for DNA repair by either homology-directed repair or non-homologous end pairing. This repaired sequence is now a part of the genomic DNA and leaves virtually no trace of gene editing. CRISPR/Cas9 gene editing has been used for several applications in oncology research and more recently to improve immune-oncology strategies. One of the earliest applications involves knockout screens in which a library of sgRNAs is used to transfect a pool of knockouts that can be used for gain-of-function or small molecule/drug screens. Genome-wide screens of cancer cell lines have been useful in identifying genes that are essential to cancer cell growth but can be prone to false-positive hits if the screens are not correctly designed or analyzed[3]. Genome-wide screens have also been used to identify genes linked to resistance, and such gene hits have been identified in a melanoma library screen when treating with the inhibitor vemurafenib[4]. Combinatorial CRISPR/Cas9 screens are now being used to target multiple genes, and this approach can be used for defining genetic or cellular pathways involved in cancer or characterizing drugs that target multiple genes[5]. CRISPR/Cas9 has moved beyond in vitro cell culture and can be used for human organoid models for studying oncogenesis. ARID1A is one of the most frequently mutated genes in solid tumors, including gastric tumors[6]. ARID1A encodes a BAF complex subunit that normally targets BAF to AT-rich DNA sequences to regulate transcription. Mouse studies have suggested that ARID1A is involved in gastrointestinal cancer oncogenesis, but this was not validated in humans until a recent study that used CRISPR/Cas9-mediated depletion of ARID1A wild-type human gastric organoids. This ARID1A-knockout organoid system established a forward genetics model of gastric cancer that resembled gastric cancer phenotypically and identified BIRC5/Survivin as a key player in early tumorigenesis. This type of organoid model is not only useful for basic oncogenesis studies but can be used for small molecule inhibitor screens. CRISPR/Cas9 screens can be used to validate mutations found in patients with gastrointestinal tumors. Activin A receptor type 2A (ACVR2A) is frequently mutated in patients with microsatellite instability-high gastric cancer, and a 2021 study used CRISPR/Cas9 to generate ACVR2A-knockout human gastric cancer cells[7]. These cells exhibited less aggressive proliferation, migration, and invasion in vitro, and patients with ACVR2A mutations had better prognoses than patients with wild-type ACVR2A. Gastrointestinal stromal tumors (GIST) have been typically characterized by activation mutations in the receptor tyrosine kinase KIT, but a 2022 study using genome-wide CRISPR screening identified the complementary chromatin-modifying complexes MOZ and Menin-MLL as being essential to GIST[8]. This finding highlights a trend in oncology research elucidating a role for chromatin regulation in tumorigenesis, which has the potential to be targeted therapeutically. Beyond basic research, CRISPR/Cas9 is starting to be used in applications for clinical trials. A Phase II trial has recently launched that is using CRISPR/Cas9 to delete an intracellular checkpoint molecule called cytokine-induced SH2 (CISH) protein[9]. Preclinical studies have shown that CRISPR/Cas9-mediated deletion of CISH enhanced antitumor responses, and the Phase II trial is using patient-derived CRISPR-modified T cells to treat individuals with metastatic gastrointestinal tumors. CAR T-cells (Chimeric Antigen Receptor) have been an incredible progress in personalized therapy targeting cancer.[10] However, different challenges hampered the availability, efficacy, and safety of this promising cell therapy. CRISPR-Cas9 technology can address many of these limitations. Desired genetic modification can be achieved with cellular precision engineering, leveraging CRISPR-Cas9-based gene editing. In addition, this approach can be exploited to overcome T-cell exhaustion, lack of CAR T-cell persistence, and toxicity related to cytokine release. In particular, the implementation of CRISPR-Cas9 technology generated universal CAR T Cells resistant to PD1 Inhibition; or enhanced T cell function against bladder cancer disrupting CTLA-4.[11] In the oncolytic therapy field, it has been demonstrated that HSV-1-derived CD40L-armed oncolytic therapy by CRISPR-Cas9-based gene editing boosted endogenous immunity in pancreatic ductal adenocarcinoma.[12] CRISPR/Cas9 gene editing will continue to transform basic and clinical research, immunotherapy, and cellular therapy providing effective and safe therapeutic strategies. Regulatory agencies are now developing frameworks to allow for the use of this revolutionary tool in a wide range of diseases. [1] Wiedenheft B, Sternberg SH, Doudna JA. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012 Feb 15;482(7385):331-8. [2] Barrangou R, Doudna JA. Applications of CRISPR technologies in research and beyond. Nat. Biotechnol. 2016;34(9):933-941. [3] Munoz DM, Cassiani PJ, Li L, Billy E, Korn JM, Jones MD, Golji J, Ruddy DA, Yu K, McAllister G, DeWeck A. CRISPR screens provide a comprehensive assessment of cancer vulnerabilities but generate false-positive hits for highly amplified genomic regions. Cancer Discovery. 2016 Aug 1;6(8):900-13. [4] Shalem O, Sanjana NE, Hartenian E, Shi X , Scott DA , et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014. 343:84–87. [5] Han K, Jeng EE, Hess GT, Morgens DW, Li A, Bassik MC. Synergistic drug combinations for cancer identified in a CRISPR screen for pairwise genetic interactions. Nat. Biotechnol. 2017 May;35(5):463-74. [6] Lo YH, Kolahi KS, Du Y, Chang CY, Krokhotin A, Nair A, Sobba WD, Karlsson K, Jones SJ, Longacre TA, Mah AT, Tercan B, Sockell A, Xu H, Seoane JA, Chen J, Shmulevich I, Weissman JS, Curtis C, Califano A, Fu H, Crabtree GR, Kuo CJ. A CRISPR/Cas9-Engineered ARID1A-Deficient Human Gastric Cancer Organoid Model Reveals Essential and Nonessential Modes of Oncogenic Transformation. Cancer Discov. 2021 Jun;11(6):1562-1581. [7] Yuza K, Nagahashi M, Ichikawa H, Hanyu T, Nakajima M, Shimada Y, Ishikawa T, Sakata J, Takeuchi S, Okuda S, Matsuda Y, Abe M, Sakimura K, Takabe K, Wakai T. Activin a Receptor Type 2A Mutation Affects the Tumor Biology of Microsatellite Instability-High Gastric Cancer. J. Gastrointest. Surg. 2021 Jan 8. doi: 10.1007/s11605-020-04889-9. Epub ahead of print. [8] Hemming ML, Benson MR, Loycano MA, Anderson JA, Andersen JL, Taddei ML, Krivtsov AV, Aubrey BJ, Cutler JA, Hatton C, Sicinska E. MOZ and Menin-MLL Complexes are Complementary Regulators of Chromatin Association and Transcriptional Output in Gastrointestinal Stromal Tumor. Cancer Discovery. 2022 May 2. [9] https://twin-cities.umn.edu/news-events/university-minnesota-opens-first-its-kind-clinical-trial-treat-metastatic-gi-cancers [10] Jogalekar MP, Rajendran RL, Khan F, Dmello C, Gangadaran P, Ahn BC.Front Immunol. 2022 Jul 22;13:925985. doi: 10.3389/fimmu.2022.925985. eCollection 2022. [11] Zhang W, Shi L, Zhao Z, Du P, Ye X, Li D, Cai Z, Han J, Cai J. Disruption of CTLA-4 expression on peripheral blood CD8 + T cell enhances anti-tumor efficacy in bladder cancer. Cancer Chemother Pharmacol. 2019;83:911–20. [12] Wang R, Chen J, Wang W, Zhao Z, Wang H, Liu S, et al. CD40L-armed oncolytic herpes simplex virus suppresses pancreatic ductal adenocarcinoma by facilitating the tumor microenvironment favorable to cytotoxic T cell response in the syngeneic mouse model. J Immunother Cancer. 2022;10(1):e003809.
Receptor Occupancy & Flow Cytometry
The development of new oncology treatments has focused on strategies that alter immune responses. Many of these novel therapies use antibodies that bind to receptors on different immune cell subsets and either activate or suppress their functions depending on the immune response being targeted. Antibody-drug conjugates are also becoming more widely used for improved targeting of drugs to specific cell subsets. Flow cytometry-based receptor occupancy (RO) assays are a valuable tool for quantifying cell surface expression of target molecules and are used to generate pharmacodynamic (PD) data, which can be used with pharmacokinetic (PK) data to guide dosing strategies. Consider these different types of receptor occupancy assays as you develop the most useful tools for screening therapeutic molecules. Total receptor assay: The evaluation of a potential therapeutic target usually begins with an assessment of how many receptors are expressed by a given cell subset. If a receptor is abundantly expressed, it may be a difficult target to use due to potential off-target effects or the requirement of high doses of a therapeutic antibody. Measurement of total receptor involves using a fluorescently labeled antibody that binds to a receptor at a site that is different from the binding site of the therapeutic antibody of interest. Free receptor assay: In this assay, cells are first stained with an unlabeled therapeutic antibody and then stained with a labeled version of the same antibody, which will only bind to the remaining free receptors. Measurement of free receptor levels at different doses is required for generating PK/PD datasets. Drug-occupied receptor assay: This assay labels cells with your antibody of interest (“drug”), and then a labeled secondary antibody binds to a different site on your antibody of interest. This measurement can be used in place of free receptor assays when they are not technically feasible. Combining data from free and total receptor assays over time is a useful method for measuring the kinetics of antibody binding and calculating potential half-lives of these molecules for therapeutic applications. Consider the type of assay you need for your phase of development. Engage with RO experts who can help you develop validated assays that can be used in preclinical and clinical settings. Flow cytometry-based RO assays will continue to be an essential tool in the development of novel oncology therapeutics.
Understanding Relapse & Acquired Resistance to CD19-targeted Therapies
Recent advances in immunotherapy have led to the development of targeted therapies for treating lymphoma and leukemia. One such therapy is CD19-targeted therapy, which is widely used as a frontline treatment for B cell lymphoma and leukemia. However, despite many patients initially respond well to this therapy relapse remains the major obstacle to be addressed. In this blog post, we will explore the various mechanisms underlying relapse to CD19-targeted therapies in patients with B cell lymphoma and leukemia. Antigen-positive Relapse: Early relapse is usually associated with short CAR-T cell persistence. This can be due to poor T cell quality, but, more importantly, it is determined by the costimulatory domain in the CAR. In fact, domains such as 4-1BB have been proven to significantly prolong the persistence of CAR-T cells[1]. In addition, the gene editing technology used to engineer the T cells can make a difference in CAR-T cell persistence. Combination therapies with checkpoint inhibitors have also been shown to prolong response duration and remission[1,2]. Immune Evasion Mechanisms: One of the primary mechanisms of acquired resistance to CD19-targeted therapies is immune evasion. Cancer cells can develop various mechanisms to evade immune surveillance and avoid being targeted by the therapy. For example, cancer cells may downregulate the expression of CD19 on their surface, thereby reducing the effectiveness of the therapy. Alternatively, they may upregulate other immune checkpoint molecules or express inhibitory proteins that prevent immune cells from recognizing and eliminating them[3]. Antigen Loss Mechanisms: Another mechanism of acquired resistance is antigen loss. CD19-targeted therapies are designed to bind to and eliminate cancer cells that express the CD19 antigen. However, cancer cells can develop mutations that lead to loss of CD19 expression, rendering the therapy ineffective. This mechanism of resistance has been observed in both lymphoma and leukemia patients, and it is a significant challenge in the development of novel therapies[4]. Development of Alternative Pathways: Cancer cells that survive CD19-targeted therapies can develop alternative pathways for survival and growth. These pathways can bypass the effects of the therapy and allow cancer cells to continue to proliferate. Examples of such mechanisms include the activation of alternative signaling pathways and the upregulation of survival factors. Understanding these adaptive mechanisms is crucial in devising new therapeutic strategies to overcome acquired resistance[1,3]. Tumor Microenvironment: The tumor microenvironment (TME) plays a critical role in acquired resistance to CD19-targeted therapies. The TME is a complex milieu of immune and stromal cells that interact with cancer cells to promote tumor growth and survival. The TME can also influence the response of cancer cells to therapy. For example, the presence of immunosuppressive cells in the TME can reduce the effectiveness of CD19-targeted therapies[1,5]. Genetic Factors: Genetic factors also play a role in acquired resistance to CD19-targeted therapies. For example, mutations in genes involved in DNA repair pathways can increase genomic instability and promote drug resistance. Identifying genetic factors that contribute to resistance can help to personalize therapy and improve outcomes[6]. Disease relapse and development of acquired resistance to CD19-targeted therapies in patients with B cell lymphoma and leukemia pose a significant clinical challenge. To overcome this challenge, we need to find ways to optimize CAR-T cell design to produce more effective therapies and understand the various mechanisms of resistance to be able to develop new therapeutic strategies that target these mechanisms. Research efforts are underway to identify novel therapeutic targets and personalized treatment approaches that can overcome acquired resistance and improve outcomes for patients[7]. Continuous improvements in our understanding of acquired resistance and relapse to CD19-targeted therapies will pave the way for the development of more effective and durable treatments for these diseases. To overcome this challenge, access to relevant preclinical models is key to develop alternative or synergistic therapeutic approaches. Champions Oncology offers a large cohort of B-cell lymphoma and leukemia models, including primary samples from patients who progressed after CD19-targeted therapies, to accelerate the development of novel therapeutic approaches to improve B-cell lymphoma and leukemia patients' lives. Shah NN, et al. Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol. 2019 Jun;16(6):372-385. doi: 10.1038/s41571-019-0184-6. PMID: 30837712; PMCID: PMC8214555. Sterner RC, et al. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 2021 Apr 6;11(4):69. doi: 10.1038/s41408-021-00459-7. PMID: 33824268; PMCID: PMC8024391. Plaks V, et al. CD19 target evasion as a mechanism of relapse in large B-cell lymphoma treated with axicabtagene ciloleucel. Blood. 2021 Sep 23;138(12):1081-1085. doi: 10.1182/blood.2021010930. PMID: 34041526; PMCID: PMC8462361. Sworder BJ, et al. Determinants of resistance to engineered T cell therapies targeting CD19 in large B cell lymphomas. Cancer Cell. 2023 Jan 9;41(1):210-225.e5. doi: 10.1016/j.ccell.2022.12.005. Epub 2022 Dec 29. PMID: 36584673; PMCID: PMC10010070. Frey NV, et al. Optimizing Chimeric Antigen Receptor T-Cell Therapy for Adults With Acute Lymphoblastic Leukemia. J Clin Oncol. 2020 Feb 10;38(5):415-422. doi: 10.1200/JCO.19.01892. Epub 2019 Dec 9. PMID: 31815579; PMCID: PMC8312030. Chen GM, et al. Characterization of Leukemic Resistance to CD19-Targeted CAR T-cell Therapy through Deep Genomic Sequencing. Cancer Immunol Res. 2023 Jan 3;11(1):13-19. doi: 10.1158/2326-6066.CIR-22-0095. PMID: 36255409; PMCID: PMC9808313. Atilla PA, et al. Resistance against anti-CD19 and anti-BCMA CAR T cells: Recent advances and coping strategies. Transl Oncol. 2022 Aug;22:101459. doi: 10.1016/j.tranon.2022.101459. Epub 2022 May 23. PMID: 35617812; PMCID: PMC9136177.

Next-Generation Sequencing Applications for Preclinical Oncology Research
Advances in preclinical oncology research are dependent on gaining insights into tumor biology and applying these insights to the development of novel diagnostics or therapeutics. Next-generation sequencing (NGS) technology has been instrumental in bridging basic immuno-oncology findings and preclinical applications. Here we provide an overview of NGS applications that are transforming preclinical oncology research. Teasing Apart the Tumor Microenvironment Solid tumors are now understood to have dynamic and unique tumor microenvironments (TMEs) that influence the immune response as well as tumor progression. Combined with flow cytometry and immunohistochemistry, single-cell NGS methods such as scRNA sequencing (scRNA-seq) and Cellular Indexing of Transcriptomes and Epitopes by Sequencing (CITE-Seq) allow scientists to measure and understand which cells are present in the TME. These findings may characterize how different stromal cells are present and contributing to tumorigenesis[1] and how the TME can create an immunosuppressive environment[2]. Elevating Liquid Biopsies Liquid biopsies have gained popularity as a method to measure circulating tumor cells (CTCs) related to either solid tumor metastasis or hematological malignancies. NGS methods have greatly improved insights gained from this sampling method[3]. Previous bulk sampling methods for quantifying CTCs were hampered by sample contamination with leukocytes and the extremely low frequency of CTCs. Single-cell NGS methods, including methods to measure single nucleotide variations as well as large-scale mutations such as copy number variations or chromosomal rearrangements, have provided unprecedented information about CTC specimens in different anatomical locations or over time. These methods have also driven the development of liquid biopsy methods that can be used to track treatment efficacy and the emergence of resistance. Better Biomarkers Tumor mutation burden (TMB) is a measurement of mutations within a tumor’s genomic sequence and has emerged as a potential biomarker associated with immune checkpoint inhibitor (ICI) efficacy. Numerous studies have now applied NGS methods to measure TMB in relation to ICI efficacy[4], and robust methods are now being validated for use clinically[5]. These findings have consistently found that high TMB correlates with ICI efficacy, and other NGS-based studies have been used to compare TMB with other genomic aberrations like microsatellite instability[6]. NGS data will continue to provide critical insights into preclinical oncology research and is certain to drive the development of better diagnostics and therapeutics. [1] Lambrechts D, Wauters E, Boeckx B, et al. (2018). Phenotype molding of stromal cells in the lung tumor microenvironment. Nature Medicine, 24(8), 1277-1289. [2] Devalaraja S, To TK, Folkert IW, et al. (2020). Tumor-derived retinoic acid regulates intratumoral monocyte differentiation to promote immune suppression. Cell.180(6):1098-114. [3] Lim SB, Di Lee W, Vasudevan J, et al. (2019). Liquid biopsy: one cell at a time. NPJ Precision Oncology. 3(1):1-9. [4] Goodman AM, Kato S, Bazhenova L, et al. (2017). Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol Cancer Ther. 16(11):2598-2608. [5] Fenizia F, Alborelli I, Costa JL, et al. (2021). Validation of a Targeted Next-Generation Sequencing Panel for Tumor Mutation Burden Analysis: Results from the Onconetwork Immuno-Oncology Consortium. J. Mol. Diagn. S1525-1578(21)00117-3. [6] Goodman AM, Sokol ES, Frampton GM, et al. Microsatellite-Stable Tumors with High Mutational Burden Benefit from Immunotherapy. (2019). Cancer Immunol. Res. 10:1570-1573.
.jpg)
Are you in the Matrix? Using Matrix vs Matrix-free assays in ex vivo culture platforms
TumorGraft3D models are Champions’ three-dimensional (3D) ex vivo tissue cultures that mimic the structure and function of tumors in vivo. They are innovative tools for studying tissue physiology, pathophysiology, and drug discovery. These models can be derived from various tissues, such as the brain, liver, intestine, etc., and can self-organize into complex structures resembling their respective tumor locations and are cultured in a matrix-free environment. The establishment and growth of 3D tissue culture models require the models to interact with the myriad of cellular and structural components within the tumor microenvironment. In this blog, we will explore the differences between matrix and matrix-free conditions in 3D tissue cultures. Traditionally, 3D tissue culture models have been cultured using a matrix-based approach, where the supportive extracellular matrix (ECM) is derived from natural or synthetic sources. Commonly used matrices include Matrigel, laminin, collagen, and hyaluronic acid. Matrigel is the most commonly used matrix for 3D tissue cultures due to its ability to mimic the natural basement membrane of the tissue. However, using matrices can also pose several challenges, such as batch-to-batch variability, limited scalability, high costs, and difficulty in downstream analysis. Additionally, animal-derived matrices, such as Matrigel, can introduce xenobiotic components that may impact the reproducibility and translatability of the results across the different species. Matrix-free approaches, on the other hand, offer an alternative to matrix-based assays. These approaches allow the self-organization of 3D tissue culture models without the use of an exogenous matrix. Matrix-free approaches are based on the principle that extracellular matrix components can be provided by the models themselves, where the cellular component of the 3D structure secretes and organizes the matrix components in a more physiological way generating a supportive microenvironment. There are several matrix-free approaches available, including hanging drop and rotary culture. These approaches offer several advantages over matrix-based assays, such as scalability, reproducibility, and compatibility with downstream analysis. In addition, this technique overcomes the species-specific differences between the cells and the matrix which will negatively influence the human translatability of the findings. Hanging drop cultures involve the formation of 3D tissue culture models in a droplet of culture medium suspended upside down. This method provides a low-adhesion environment for the models to organize and secrete their own matrix components. Rotary culture is another matrix-free approach that involves the constant rotation of a culture vessel, allowing the models to grow and form a 3D structure without the need for a supporting matrix. Both matrix-based and matrix-free assays have pros and cons for 3D tissue cultures. Matrix-based assays mimic the ECM of the tissue providing an artificial microenvironment for model growth. Beyond batch-to-batch variability, limited scalability, and high costs; including matrix in your ex vivo cultures can also represent a biological problem. Given the artificial architecture of the matrix, it imposes unnatural stiffness and pre-defined structures that will influence the hierarchical organization of the cells during 3D structure generation. Matrix-free assays offer an alternative that eliminates the need for exogenous matrix components and provides a low-adhesion environment for the 3D tissue culture growth for a fully human and more physiological microenvironment. While matrix-free approaches are promising, they are not without their challenges. Specifically, there is the need for ad-hoc development of various protocols for each different tumor type, including rigorous procedures for optimization and standardization to ensure results are reproducible. In summary, the decision to use matrix or matrix-free culture conditions in 3D tissue culture assays can influence the downstream translatability of your assay. Researchers should evaluate the differences and understand how using a matrix or matrix-free culture conditions can impact their research. References: 1) Loewa, A., Feng, J.J. & Hedtrich, S. Human disease models in drug development. Nat Rev Bioeng (2023). https://doi.org/10.1038/s44222-023-00063-3 2) Proietto M, Crippa M, Damiani C, Pasquale V, Sacco E, Vanoni M, Gilardi M. Tumor heterogeneity: preclinical models, emerging technologies, and future applications. Front Oncol. 2023 Apr 28;13:1164535. doi: 10.3389/fonc.2023.1164535. PMID: 37188201; PMCID: PMC10175698. 3) LeSavage BL, Suhar RA, Broguiere N, Lutolf MP, Heilshorn SC. Next-generation cancer organoids. Nat Mater. 2022 Feb;21(2):143-159. doi: 10.1038/s41563-021-01057-5. Epub 2021 Aug 12. PMID: 34385685. 4) Kozlowski MT, Crook CJ, Ku HT. Towards organoid culture without Matrigel. Commun Biol. 2021 Dec 10;4(1):1387. doi: 10.1038/s42003-021-02910-8. PMID: 34893703; PMCID: PMC8664924.

Top 3 Considerations for Using Ex Vivo Hematologic Models
Advances in precision medicine have transformed treatments for many types of solid tumors, but similar treatment options have been more limited for hematologic oncology. Now, new ex vivo hematologic oncology models are being developed that use patient-derived lymphoma or leukemia cells for screening experimental drugs or biologics. The lower cost associated with ex vivo drug screening studies compared to in vivo ones makes these platforms ideal for large scale screening of therapeutic agents before progressing to in vivo studies with the most promising drug candidates. In addition, ex vivo hematologic oncology models closely recapitulate the tumor of origin and can provide valuable information about the variability of drug response in a cohort of patients. In expert hands, these platforms are well-suited to preclinical drug screening and can inform preclinical research decisions as well as clinical care. Some unique aspects of ex vivo hematologic oncology models need to be considered as you advance your research. Viability, Survival, and Proliferation: Patient-derived ex vivo hematologic oncology models may adapt to culture conditions very differently; some cells may die off significantly whereas others proliferate at high rates. It is critical to measure and understand the proliferation and viability parameters of your ex vivo hematologic oncology models to assure you are working with a reliable specimen. Consider Specific Cell Characteristics: Prolonged culturing of ex vivo hematologic oncology models may introduce unwanted changes to cells that can alter their responses to drugs or antibodies. Be sure to thoroughly characterize the phenotype of specific ex vivo hematologic oncology models before and during ex vivo culture to assure that prolonged culture does not significantly alter the cell phenotype or cause unintended mutations. Short term cultures reduce the risk of such changes happening. The Power of the Panel: A major advantage of using ex vivo hematologic oncology models is the ability to scale up cultures and screen large panels of experimental antibodies or drugs. This can also be done with multiple patient-derived ex vivo hematologic oncology models being screened in parallel. Ex vivo hematologic oncology models have advanced significantly as ex vivo drug screening platforms have emerged. With our expertise and our large bank of ex vivo hematologic oncology models, at Champions Oncology, we are well poised to help you advance your preclinical therapeutic screening. Leveraging the multi-omics and multimodal characterization of our ex vivo hematologic oncology models will help you narrow down your drug candidates and speed up the trajectory to clinical investigation.

The Dos & Don’ts of Flow Cytometry in the Clinic
Clinical flow cytometry is now a standard tool used by clinical researchers in numerous fields, especially immuno-oncology. Consider these “Dos & Don’ts” of clinical flow cytometry as you decide to use this tool for clinical research. Do Perfect Your Panel - A staining panel includes the set of antibodies used to identify different cell subsets within a sample and depends on the use of multiple antibodies conjugated with different fluorochromes. These fluorochromes are excited by specific light wavelengths from the cytometer’s lasers, and the emitted light is analyzed and interpreted as different cell types. Researchers take time to develop staining panels that can reasonably distinguish between cell types and avoid aberrations such as spectral overlap. Do Vigilant Validation - Clinical flow cytometry assay development must satisfy certain criteria, including accuracy, specificity, limit of detection, reproducibility, precision, linearity, and stability. By satisfying these criteria, researchers are able to have confidence in their clinical data, even if experiments were done at different times or in different locations. Do Maintenance - Reliable flow cytometry data requires a well-designed and validated protocol as well as a properly maintained cytometer. Routine maintenance is carried out by cytometer users to assure that fluidic systems flow properly, lasers are firing correctly, and the emitted light is detected at the expected wavelength. Infrequent, but more involved, maintenance is carried out by technicians and is essential to the long-term functionality of a cytometer. Don’t Pick the Wrong Test Tube - The very first step of a clinical flow cytometry protocol is collecting a sample, which is typically peripheral blood, cord blood, or tissue biopsy. These samples can be collected in tubes with preservatives or anti-clotting agents, but some of these additives can alter cell viability. It is worthwhile to evaluate different sample tube types to assure that your cells of interest are not being lost or damaged due to test tube effects. Don’t Lose your Cells - Clinical flow cytometry protocols rely on the acquisition of thousands to millions of cells per sample in order to have a reasonable number of events to analyze. One essential step to acquiring usable data is the inclusion of a viability dye that discriminates between live and dead cells. Viability dyes come in different formats that work with specific flow cytometer configurations, so it is critical to determine which dye will work with your cytometer. Don’t Screw Up Your Analysis - After you have completed the laborious process of sample staining and data acquisition on a cytometer, it is essential to execute the appropriate data analysis. Be sure you understand how to use the software needed for analysis, and this may mean including certain controls in your staining and acquisition protocols. Errors in analysis may result in your data being inaccurate or undetected. There are many considerations needed for a clinical flow cytometry protocol, and it may be worthwhile to work with experts in protocol development, assay validation, or general clinical flow cytometry.
Insights in Invasive Bladder Cancer
Bladder cancer is a relatively common form of cancer that is defined as either pre-invasive or invasive, and non-muscle invasive bladder cancer (NMIBC) is the most commonly diagnosed subtype[1]. NMIBC is typically treated by surgical resection and/or intravesical delivery of chemo- or immunotherapy-based adjuvant treatment, and long-term efficacy is monitored by urine testing or cystoscopy. Muscle-invasive bladder cancer (MIBC) is relatively resistant to current treatment options and occurs more frequently in men. MIBC also has high rates of morbidity and mortality, and novel therapies or combination therapies are being developed to better treat this form of bladder cancer. Here we highlight recent findings about invasive bladder cancer biology and how these observations are informing the development of new therapies. Mutation Landscape Analysis of MIBC genomes has revealed a high mutation rate in tumors that approaches rates seen in melanoma and lung cancer[2]. The mutational signature of many MIBC tumors is consistent with mutations associated with APOBEC (apolipoprotein B mRNA-editing enzyme, catalytic polypeptide-like) cytidine deaminase, and many of the mutations are clonal, which suggests that the effects of APOBEC expression in bladder tissue occurs early in tumorigenesis. Non-invasive and invasive bladder tumors are also often classified as being highly heterogeneous, and tumor histology can be extremely variable and includes squamous, plasmacytoid, micropapillary, and small-cell carcinoma. This molecular and histological heterogeneity presents a challenge in subtyping tumors based on gene expression profiles and provides insight into why it can be difficult to identify targeted therapies that are effective against bladder cancers. Several genes are commonly mutated in both NMIBC and MIBC, but mutation rates for some of these genes, including MLL2 and TP53, are higher in invasive tumors[3]. TP53 mutations appear to be a critical factor associated with the transition to its invasive form. Mutations have also been identified in genes associated with the sister chromatid cohesion and segregation process (STAG2, ESPL1, FGFR3, and TACC3)[3] and chromatin remodeling (UTX, MLL-MLL3, CREBBP-EP300, NCOR1, ARID1A, and CHD6)[4]. Immunotherapy Insights NIMBC is one of the first forms of cancer treated with an immunotherapy approach in which the attenuated tuberculosis bacterial vaccine, Bacillus Calmette-Guérin (BCG), is delivered intravesically to the bladder and triggers an immune response that inhibits tumor growth[5]. Recently, immune checkpoint inhibitor-based treatments have been examined as therapeutic options for different bladder cancers. Expression of the PD-L1 immune checkpoint molecule has been detected on both NIMBC and MIBC tumor cells and tumor-infiltrating mononuclear cells. Antibody-based blockade of PD-1/PD-L1 interactions restores anti-tumor responses mediated by T cells. Atezolizumab was the first PD-L1 inhibitor approved for the treatment of locally advanced/ metastatic urothelial carcinoma (LA/mUC) that has progressed or after cisplatin-based chemotherapy[6], and five different PD-L1/PD-1 inhibitors have been approved for this form of bladder cancer. Other checkpoint inhibitors that target different molecules have not shown significant improvements in overall survival or progression-free survival. Clinical trials currently underway are examining the efficacy of combinations of checkpoint inhibitors or the use of these inhibitors as an adjuvant therapy to surgical resection or chemotherapy. In addition, therapies that target specific mutations, such as FGFR, HER2, or chromatin-modifying genes, are also under investigation, and the FGFR inhibitor erdafitinib was recently approved for the treatment of LA/mUC[7][8]. Bladder cancer treatment outcomes continue to advance because of insights into the basic biology of bladder cancer, and a better understanding of molecular and immunological mechanisms associated with anti-tumor responses. The many preclinical and clinical studies currently underway for bladder cancer will contribute valuable information to new therapeutic options, especially for patients with refractory or invasive forms of bladder cancer. [1] Knowles MA, Hurst CD. Molecular Biology of Bladder Cancer: New Insights into Pathogenesis and Clinical Diversity. Nat Rev Cancer. 2015;15:25–41. [2] Robertson AG, Kim J, Al-Ahmadie H, et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell. 2017;171(3):540-556.e25. [3] Guo G, Sun X, Chen C, et al. Whole-Genome and Whole-Exome Sequencing of Bladder Cancer Identifies Frequent Alterations in Genes Involved in Sister Chromatid Cohesion and Segregation. Nat Genet. 2013;45(12):1459-1463. [4] Gui Y, Guo G, Huang Y, et al. Frequent Mutations of Chromatin Remodeling Genes in Transitional Cell Carcinoma of the Bladder. Nat Genet. 2011;43(9):875-878. [5] Herr HW and Morales A. History of Bacillus Calmette-Guerin and Bladder Cancer: An Immunotherapy Success Story. J Urol. 2008;179.1: 53-56. [6] Bellmunt J, Powles T, Vogelzang NJ. A Review on the Evolution of PD-1/PD-L1 Immunotherapy for Bladder Cancer: The Future is Now. Cancer Treat. Rev. 2017;54:58-67. [7] Osterman CK, Milowsky MI. New and Emerging Therapies in the Management of Bladder Cancer. F1000Res. 2020;9:F1000 Faculty Rev-1146. 1 [8] Loriot Y, Necchi A, Park SH, et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N Engl J Med. 2019 Jul 25;381(4):338-348.
Head & Neck Cancers - New Mutation Insights Lead to New Therapies
Head and neck squamous cell carcinoma (HNSCC) is one of the most common forms of cancer in the world. This heterogeneous disease is most often seen in men and is closely linked to tobacco usage, which can be enhanced by alcohol usage. Human papillomavirus (HPV) infection is an independent risk factor for HNSCC[1], and most HPV- HNSCCs occur in the larynx and oral cavity, and HPV+ HNSCCs typically arise in the oropharynx[2]. Both HPV+ and HPV- HNSCCs are typically diagnosed at advanced disease stages and are often treated with an aggressive combination of surgery, radiotherapy (RT), and chemotherapy[3]. More recently, cetuximab, an IgG1 monoclonal antibody that targets epidermal growth factor receptor (EGFR) has been used in combination with radiotherapy and has shown some improvements in progression-free survival (PFS) and overall survival (OS)[4]. Advances in genomic technology have provided critical insights into the pathology of HNSCC subtypes. Some HNSCCs are now known to have high mutational burdens, which is like other cancers associated with carcinogen exposure. Analysis of HPV- and HPV+ HNSCCs in The Cancer Genome Atlas (TGCA) network has been instrumental in identifying genome defects common to both HPV+ and HPV- HNSCCs, as well as defects that are unique to each of these subtypes. Global analysis of all HNSCC subtypes identified TP53 as the most commonly mutated gene, but there tends to be a lower percentage of TP53 mutations in HPV+ tumors versus HPV- tumors, likely related to HPV infection[5]. Activating mutations in PIK3CA and inactivating mutations in NOTCH1 are associated with both types of HNSCC. HPV- HNSCCs show inactivating mutations in TP53 and CDKN2A cell cycle suppressor genes and amplification of CCND1, which can all contribute to unchecked cell cycle progression. In contrast, HPV+ HNSCCs show expression of the E6 and E7 viral oncogenes, which can inhibit TP53 and RB1, as well as activation of cell cycle regulator E2F1[6]. Mutations in the MAPK signaling component HRAS have also been identified in a fraction of HNSCC specimens for which treatment options are limited. A recent clinical trial demonstrated encouraging efficacy in patients with recurring or metastatic HNSCCs carrying HRAS mutations[7] treated with tipifarnib, which is a farnesyltransferase inhibitor that can inhibit farnesylation of HRAS and membrane targeting and downstream signaling. In addition to targeted therapies, recent studies suggest that PD-1/PD-L1 blockade alone or in combination with chemotherapy can improve overall survival in patients with recurrent or metastatic HNSCC[8],[9]. PD-1 blockade has shown better survival, toxicity, and response duration compared with cetuximab treatment combined with chemotherapy, but these responses are dependent on patients having HNSCC tumors that are positive for PD-L1. Tumor mutational burden and the expression of inflammatory biomarkers also correlated directly with responses to PD-1 blockade. Recent insights into genomic variations associated with HNSCC subtypes are providing valuable information with respect to basic tumor biology as well as identifying potential therapeutic targets for preclinical development. [1] Cognetti DM, Weber RS, Lai SY. Head and neck cancer: an evolving treatment paradigm. Cancer. 113(7 Suppl):1911-1932 (2008). [2] Mehanna H, Beech T, Nicholson T, El-Hariry I, McConkey C, Paleri V, Roberts S. Prevalence of human papillomavirus in oropharyngeal and nonoropharyngeal head and neck cancer--systematic review and meta-analysis of trends by time and region. Head Neck. 35(5):747–55 (2013). [3] Pignon J P, le Maître A, Maillard E, Bourhis J. & MACH-NC Collaborative Group. Meta-analysis of chemotherapy in head and neck cancer (MACH-NC): An update on 93 randomized trials and 17,346 patients. Radiother. Oncol. 92(1), 4–14 (2009). [4] Bonner JA, et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomized trial, and the relation between cetuximab-induced rash and survival. Lancet Oncol. 11, 21–28 (2010). 5] Westra, W. H. et al. The inverse relationship between human papillomavirus-16 infection and disruptive p53 gene mutations in squamous cell carcinoma of the head and neck. Clin. Cancer Res. 14, 366–369 (2008) [6] Beck TN, Golemis EA. Genomic insights into head and neck cancer. Cancers Head Neck 1, 1 (2016). [7] Ho AL, Brana I, Haddad R. et al. Tipifarnib in Head and Neck Squamous Cell Carcinoma with HRAS Mutations. J. Clin. Oncol. JCO2002903 (2021). [8] de Sousa LG, Ferrarotto R. Pembrolizumab in the first-line treatment of advanced head and neck cancer. Expt. Rev. Anticanc. Ther. 21.12: 1321-1331 (2021). [9] Haddad RI, Seiwert TY, Chow LQM et al. Influence of tumor mutational burden, inflammatory gene expression profile, and PD-L1 expression on response to pembrolizumab in head and neck squamous cell carcinoma. J. Immunother. Cancer. 2022 Feb;10(2):e003026.
Novel RNAs Insights Toward AML Development
Acute myeloid leukemia (AML) is an aggressive hematological malignancy and the most common leukemia among the adult population. Despite the development of novel targeted therapies, resistance to treatments and disease relapse remain unsolved. New interest in the role of circular RNA in AML biology has opened the way for the development of new approaches in the management of AML. Somatic mutations in the additional sex comb-like 1 (ASXL1) gene have been identified in multiple hematologic malignancies, including acute myeloid leukemia (AML), and are associated with poor prognoses1. ASXL1 encodes a nuclear protein that regulates epigenetic remodeling and transcription through interactions with polycomb complex proteins and transcriptional activators and repressors. Several ASXL1 mutations have been associated with loss of protein expression that leads to myeloid transformation2. In contrast, gain-of-function mutations in ASXL1 result in expression of a truncated ASXL1 protein that can bind to BRCA-1 associated protein 1 (BAP1) and cause leukemogenesis3,4. Targeted reduction of BAP1 activity is sufficient to prevent this malignancy process5. A recent study has shown that the ASXL1 gene locus undergoes alternative splicing to produce circular RNAs (circRNAs) in addition to linear protein-coding mRNAs6. CircRNAs are non-coding RNAs that normally function as regulators of gene expression and translation by acting as sponges for microRNAs or forming complexes with RNA-binding proteins. Here they identified two isoforms of circular ASXL1 (circASXL1), and they showed that circASXL1-1 can bind to BAP-1 and regulate BAP1-mediated deubiquitinase activity, which targets H2AK119 ubiquitination and regulates myeloid differentiation of hematopoietic stem cells. These findings suggest that circRNAs may be developed as novel therapeutics for treating hematologic malignancies like AML. Pratcorona M, Abbas S, Sanders MA, et al. Acquired mutations in ASXL1 in acute myeloid leukemia: prevalence and prognostic value. Haematologica. 2012;97(3):388-392. Abdel-Wahab O, Adli M, LaFave LM, et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell. 2012;22(2):180-193. Balasubramani A, Larjo A, Bassein JA, et al. Cancer-associated ASXL1 mutations may act as gain-of-function mutations of the ASXL1-BAP1 complex. Nat Commun. 2015;6:7307. Asada S, Goyama S, Inoue D, et al. Mutant ASXL1 cooperates with BAP1 to promote myeloid leukaemogenesis. Nat Commun. 2018;9(1):2733 Guo Y, Yang H, Chen S, et al. Reduced BAP1 activity prevents ASXL1 truncation-driven myeloid malignancy in vivo. Leukemia. 2018;32(8):1834-1837. Jadhav SP, Kumari N, Ng L, et al. circASXL1-1 regulates BAP1 deubiquitinase activity in leukemia. Haematologica, 2020; 105 (7): e343 DOI: 10.3324/haematol.2019.225961.
Overcoming Drug Resistance in AML: Targeting Tricky Signaling Pathways
Resistance to anti-cancer treatments is a significant therapeutic challenge and is typically associated with mutations in signaling pathways that are involved in cell proliferation, survival, and tumorigenesis. Genomic analyses of relapsed pediatric acute myeloid leukemia (AML) patients have identified mutations, deletions, and changes in promoter methylation associated with Wnt-β-catenin signaling[1],[2]. Mutations in the PI3K-Akt pathway are also frequently associated with treatment resistance in AML and many other cancers[3],[4]. Hematopoietic stem cells are particularly sensitive to mutations in these signaling pathways and numerous studies have shown that targeting each of these pathways separately is associated with poor efficacy and the emergence of resistance[5]. AML resistance not only emerges from chemotherapy but can also be seen in response to immunotherapy[6]. Combined therapies are emerging as a strategy to overcome treatment resistance. Numerous clinical trials are examining the efficacy of combining chemotherapy and immunotherapy-based approaches or using differing immunotherapy combinations to enhance anti-tumor immunity. Other studies are seeking to use existing drugs in different ways to avoid resistance. A recent study has shown that the anthracycline antibiotic doxorubicin (DXR) can be repurposed as a β-catenin inhibitor that targets leukemia stem cells (LSCs)[7]. DXR has typically been used as a chemotherapeutic agent at high doses but is associated with significant toxicity. In this study, activation of both the Wnt-β-catenin and PI3K-Akt pathways are associated with the expansion of LSCs. A high throughput screening of a small molecule library identified DXR as an inhibitor of β-catenin activation mediated by interactions with Akt, and low-dose DXR treatment was sufficient to inhibit LSC expansion, even in chemo-resistant pS552-β-cat+ LSCs. DXR could also reduce the expression of the immune checkpoint molecules PD-L1, TIM3, and CD24 on LSCs and thus reverse immune checkpoint-mediated resistance. Findings from a pilot clinical trial indicated that low-dose treatment with the DXR analogue daunorubicin of adult patients with relapsed or refractory AML can specifically target chemo-resistant pS552-β-cat+ LSCs. Future studies using next-generation sequencing technology, computational analysis, and high throughput screening will continue to advance the development of treatments for relapsed or refractory cancers. Progress also continues to be made in advancing combined treatments for both hematological malignancies and solid tumors. 1. Hogan, L. E. et al. Integrated genomic analysis of relapsed childhood acute lymphoblastic leukemia reveals therapeutic strategies. Blood. 2011; 118: 5218–5226. 2. Bolouri, H. et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat. Med. 2018; 24: 103–112. 3. Koren, S. & Bentires-Alj, M. Tackling resistance to PI3K inhibition by targeting the epigenome. Cancer Cell. 2017; 31: 616–618. 4. Lindblad, O. et al. Aberrant activation of the PI3K/mTOR pathway promotes resistance to sorafenib in AML. Oncogene. 2016; 35: 5119–5131. 5. Fruman, D. A. & Rommel, C. PI3K and cancer: lessons, challenges and opportunities. Nat. Rev. Drug Discovery. 2014. 13; 140–156. 6. Sharma, P. et al. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017. 168; 707–723. 7. Perry, J.M. et al. Overcoming Wnt-β-catenin dependent anticancer therapy resistance in leukemia stem cells. Nat Cell Biol. 2020. 22(6); 689-700.
UnTIL We Meet Again: Testing TIL Therapies in an Ex Vivo Platform
Tumor-infiltrating lymphocyte (TIL)-based immunotherapy is currently at the forefront of cutting-edge immuno-oncology treatments. TILs are a type of adoptive cellular therapy (ACT) using lymphocytes that are found within tumor tissues; most of these lymphocytes are T cells that can specifically target tumor cells. For TIL-based therapies, these T cells are harvested from a tumor biopsy, expanded ex vivo, and infused back into the patient. Advances in TIL-based therapies are driven by preclinical characterization and screening of TILs against a wide array of tumor types. Consider these factors related to preclinical TIL therapy studies as you develop new research protocols: Wanted Alive, Not Dead: The development of plate-based TIL assays offers users flexibility and scalability, but it is critical to assess the viability of cultured TILs at the outset of any of these assays. Some TIL cultures are prone to high levels of cell death over time, and culture conditions may need to be optimized to include different cytokines and growth factors to promote viability as well as growth and expansion of tumor-specific T cells. Cell Selection: Optimal TIL culture conditions should enhance the expansion of tumor-specific T cells but may also involve a step to deplete other cells prior to expansion. This may involve the depletion of adherent cell subsets with immunosuppressive characteristics such as myeloid-derived suppressor cells (MDSCs). Removing these cells from ex vivo culture allows TILs to expand and differentiate into T cells with tumor-specific cytotoxic activity. TIL Test Drive: Ex vivo expansion of TILs must include functional assays in order to ascertain if these cells have anti-tumor properties. Analysis typically includes flow cytometry-based immunophenotyping and characterization of cytokine production and tumor-specific cytotoxicity. An ideal TIL product will show tumor-specific activity with limited off-target effects, and TILs will retain a phenotype that is predicted to be well-tolerated in a patient upon re-infusion. Preclinical TIL studies are continuing to expand the usage of TIL-based therapies against a wide range of solid tumors, and this field of study will advance further as ex vivo culture and analysis techniques improve.

Clinical Flow Cytometry – Understanding GCLP Standards and Compliance
Flow cytometry is a semi-quantitative method that is widely used for preclinical and clinical studies. Preclinical flow cytometry experiments rely on users who understand the fundamentals of setting up appropriate standards and controls, but these protocols do not need to typically meet specific clinical standards. In contrast, clinical flow cytometry protocols require adherence to good clinical laboratory practice (GCLP) standards. Here we provide an overview of GCLP standards with respect to clinical flow cytometry. GCLP guidelines were developed in the early 2000s to provide a regulatory framework for labs performing assays for HIV-1 clinical trials. A large portion of these assays were flow cytometry-based[1] and were being conducted on flow cytometers at multiple global sites, thus necessitating standardization. GCLP guidelines are a type of regulatory standard, with similarities to good laboratory practice (GLP) and clinical laboratory improvement amendments (CLIA) and have become essential to harmonizing clinical laboratory work taking place at different sites and times for clinical trials. GCLP standards can be applied to labs carrying out safety, diagnostic, or endpoint assays, and have been critical to the evaluation of immuno-oncology drugs and biologics being evaluated in clinical trials. GCLP guidelines include personnel organization and training, equipment maintenance and validation, the development and validation of standard operating procedures (SOPs), assay development and validation, and audits to identify discrepancies in any of these areas[2]. GCLP procedures for clinical flow cytometry assure that flow cytometers at different sites are performing consistently and within standards. Flow cytometry reagents are parallel tested with previous lot to assure their eligibility for use in an assay. Throughout the implementation of GCLP guidelines, a laboratory must maintain a document control plan that includes all SOPs currently in use and an archive system for SOPs that are no longer in use. A quality control (QC) program is essential to a GCLP framework as it allows for the monitoring, documentation, and recognition of QC problems. QC programs track test standards, positive and negative controls, reagent performance, QC data analysis and logs, and parallel testing. QC is especially important for clinical flow cytometry to assure that the appropriate cell populations are being analyzed in a consistent and reproducible manner. A proficiency testing program is another critical component of GCLP program because it involves analysis of a common set of samples by multiple laboratory sites to assure that performance can meet specific criteria[3]. Clinical flow cytometry assays typically lack “gold standard” samples for evaluation because cells that are being analyzed in this manner are subject to intra- and inter-sample variability. Nonetheless, samples can be expected to fall within a specific predetermined range, thus assuring that the GCLP-compliant protocol, users, and equipment are performing proficiently. GCLP standards for clinical flow cytometry are now being applied in different ways, including complex immunophenotyping of multiple immune cell subsets, comprehensive measurement of immune checkpoint molecule expression on T cells, and receptor occupancy assessment of therapeutic antibodies. The development of GCLP guidelines is often carried out with GCLP professionals who can advise on all aspects of this complex undertaking. Alternatively, individual assays, such as clinical flow cytometry experiments can be carried out by contract research organizations who are already GCLP compliant and experienced with running these assays. [1] Todd CA, Sanchez AM, Garcia A, Denny TN, Sarzotti-Kelsoe M. Implementation of Good Clinical Laboratory Practice (GCLP) guidelines within the External Quality Assurance Program Oversight Laboratory (EQAPOL). J. Immunol. Methods. 2014;409:91-98. [2] Sarzotti-Kelsoe M, Cox J, Cleland N, et al. Evaluation and recommendations on good clinical laboratory practice guidelines for phase I-III clinical trials. PLoS Med. 2009;6(5):e1000067. [3] O'Hara, Denise M., et al. Recommendations for the validation of flow cytometric testing during drug development: II assays. J. Immunol. Methods. 363.2 (2011): 120-134.
Targeted Therapy for Biliary Cancers
Biliary tract cancers, which are also known as cholangiocarcinomas (CCA), describe malignancies that occur in the biliary tract and include the pancreas, gallbladder, and bile ducts. These are relatively rare cancers but are associated with a poor prognosis given that these cancers are difficult to detect and are usually diagnosed at later stages of the disease. Current CCA classification and clinical care CCAs are classified by growth pattern as mass forming, intraductal, or periductal, anatomical location as intrahepatic or extrahepatic (perihilar, or distal), and histology (predominantly adenocarcinomas with mixed hepatocellular CCAs are defined as a separate subtype of primary hepatic cancer) [1]. CCAs can arise de novo with no discernable risk factors, but a variety of genetic and nongenetic risk factors are associated with CCA, including liver cirrhosis, obesity, type 2 diabetes, chronic hepatitis B or C infection, or infection with hepatobiliary flukes [2]. Individuals with primary sclerosing cholangitis have higher levels of chronic liver inflammation and increased incidence of CCA [3] as well. CCAs are typically aggressive cancers with a median overall survival of < 24 months. The only treatment options have been surgical resection or liver transplantation for early-stage patients or aggressive chemotherapy for patients with advanced stages of the disease [4]. CCA molecular classification Next-generation sequencing technology has allowed the identification of molecular subtypes associated with several gene mutations that are linked to increased CCA risk, including mutations in chromatin remodeling genes (ARID1A, BAP1, and PBRM1) [5,6]. IDH1 mutations have been linked to intrahepatic CCA and mutations in ERBB2 are more closely associated with extrahepatic CCA [7]. Mutations in the fibroblast growth factor (FGF) signaling pathway, including mutations or translocations in FGF receptor (FGFR) genes, have also been identified in CCA, and an FGFR fusion has been linked specifically to intrahepatic CCA [8]. Moreover, patients with IDH1/2 mutations also exhibit elevated FGFR expression, even without FGFR mutations or fusions, further affirming a role for FGF/FGFR signaling in CCA progression[9]. In addition to tumor cell alterations, the tumor microenvironment is also being extensively explored and has led to the identification of an inflamed intrahepatic CCA subtype potentially treatable with checkpoint blockade immunotherapy [10]. New CCA precision therapies The FGF/FGFR pathway has become one of the most encouraging targets for novel CCA therapeutics. As FGFRs are receptor tyrosine kinases, nonselective FGFR inhibitors that target the conserved ATP-binding domain were the first drugs to be tested in clinical trials. A recent trial exploring the use of the nonselective FGFR kinase inhibitor pazopanib combined with a MEK inhibitor showed little efficacy [11]. Pemagatinib is the first FDA-approved targeted therapy for advanced CCA with FGFR2 fusions or rearrangements. Pemagatinib is a selective inhibitor of FGFR1, 2, and 3 and was approved in 2020 and given breakthrough therapy designation as the first drug to specifically treat CCA [12]. In 2021, Ivosidenib (Tibsovo), a small molecule IDH1 inhibitor, was approved by the FDA for treating IDH1 mutated locally advanced or metastatic CCA [13]. Very recently, the FDA has granted accelerated approval to two FGFR inhibitors: Infigratinib (Truseltiq), for the treatment of previously treated, unresectable, locally advanced or metastatic cholangiocarcinoma with FGFR2 fusion (and a companion diagnostic to identify patients eligible to receive this treatment) [14,15]; and Futinatinib (TAS-120) for patients with previously treated, unresectable, locally advanced or metastatic intrahepatic CCA with FGFR2 gene fusions or other rearrangements [16,17]. Alternative FGF/FGFR inhibitors are also under development and target the extracellular domain or function as FGF ligand traps [18]. These alternative inhibitors can be used potentially in combination with FGFR kinase inhibitors that have lost efficacy due to the development of resistance [19]. In addition to mutation-driven therapeutic approaches, immune checkpoint inhibitors are currently tested in combination with chemotherapy to improve efficacy. A very recent study conducted on patients with advanced biliary tract cancer showed that Durvalumab, a PD-L1 inhibitor, significantly improves survival compared to chemotherapy alone [20]. New research on novel inhibitors and combination therapies will likely identify other therapeutic options for CCA, and these cancers will not be the terminal diagnoses they once were. Razumilava N, Gores Cholangiocarcinoma. Lancet. 2014;383(9935):2168- 79. Yao KJ, Jabbour S, et al. Increasing mortality in the United States from cholangiocarcinoma: an analysis of the National Center for Health Statistics Database. BMC Gastroenterol. 2016;16(1):117. Hirschfield GM, Karlsen TH, et Primary sclerosing cholangitis. Lancet. 2013;382(9904):1587-99. Yazici C, Niemeyer DJ, et al. Hepatocellular carcinoma and cholangiocarcinoma: an Expert Rev Gastroenterol Hepatol. 2014;8(1):63-82. Jusakul A, Cutcutache I, et al. Whole‐genome and epigenomic landscapes of etiologically distinct subtypes of cholangiocarcinoma. Cancer Discov 2017;7:1116‐1135 Jiao Y, Pawlik TM, et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A, and PBRM1 in intrahepatic Nat Genet. 2013;45:1470–3. Churi CR, Shroff R, et Mutation profiling in cholangiocarcinoma: prognostic and therapeutic implications. PLoS One. 2014;9:e115383. Graham RP, Barr Fritcher EG, et al. Fibroblast growth factor receptor 2 translocations in intrahepatic Hum Pathol. 2014;45(8):1630- 1638. Wang P, Dong Q, et Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene. 2013;32(25):3091-3100. Job S, Rapoud D, et al.. Identification of Four Immune Subtypes Characterized by Distinct Composition and Functions of Tumor Microenvironment in Intrahepatic Cholangiocarcinoma. Hepatology. 2020 Sep;72(3):965-981 Shroff RT, Yarchoan M, O’Connor A, et The oral VEGF receptor tyrosine kinase inhibitor pazopanib in combination with the MEK inhibitor trametinib in advanced cholangiocarcinoma. Br J Cancer. 2017;116(11):1402-1407. Patel TH, Marcus L, et al. FDA Approval Summary: Pemigatinib for Previously Treated, Unresectable Locally Advanced or Metastatic Cholangiocarcinoma with FGFR2 fusion or other rearrangements. Clin Cancer Res. 2022 Oct 7:CCR-22-2036. Sumbly V, Landry I, Rizzo V. Ivosidenib for IDH1 Mutant Cholangiocarcinoma: A Narrative Review. Cureus. 2022 Jan 7;14(1):e21018 Javle M, Roychowdhury S, et al. Infigratinib (BGJ398) in previously treated patients with advanced or metastatic cholangiocarcinoma with FGFR2 fusions or rearrangements: mature results from a multicentre, open-label, single-arm, phase 2 study. Lancet Gastroenterol Hepatol. 2021 Oct;6(10):803-815. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-infigratinib-metastatic-cholangiocarcinoma Goyal L, Meric-Bernstam F, et al. Futibatinib for FGFR2-Rearranged Intrahepatic Cholangiocarcinoma. N Engl J Med. 2023 Jan 19;388(3):228-239. doi: 10.1056/NEJMoa2206834. PMID: 36652354. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-futibatinib-cholangiocarcinoma Tolcher AW, Papadopoulos KP, Patnaik A, et A phase I, first in human study of FP-1039 (GSK3052230), a novel FGF ligand trap, in patients with advanced solid tumors. Ann Oncol. 2016;27(3):526-532. Touat M, Ileana E, et Targeting FGFR signaling in cancer. Clin Cancer Res. 2015;21(12):2684-2694. https://meetings.asco.org/abstracts-presentations/204876

Leveraging IHC as a powerful clinical tool for IO therapies
Immunohistochemistry (IHC) is based on antigen–antibody binding to directly visualize the status of markers of interest in tissues. IHC provides a huge amount of information and is now indispensable in many fields including pathology, cancer biology, and drug discovery. IHC has been and continues to be analyzed by pathologists using microscopy. For pathologists in everyday practice or in the research context, IHC is an essential tool to interpret the pathophysiology of diseased tissues. Furthermore, IHC is also a crucial tool for biomarker discovery validation leading to personalized medicine. It provides information about the localization and the abundance of the target protein, both within tissue and at the sub-cellular level. More recently, growing evidence of the importance of spatial information enclosed in the tissue for patient stratification leading to improved clinical outcomes further boosted the development and standardization of many different and improved image analysis methods. This is particularly true in the field of immune-oncology and checkpoint inhibitors therapeutic development, where the presence of immune cells in the tumor correlated with positive clinical outcomes in many different types of cancer. IHC Analysis and Scoring Methods IHC results can be variable in both the percentage of positive detected cells as well as in protein expression intensity. The spatial distribution of the staining is also very variable. Sometimes the staining is widely spread out across the tissue while other times it is localized in small specific areas. The heterogeneity of the tissues reflected in the IHC staining needs to be classified, and more standardized scoring methods were needed to use this powerful tool in the clinic to stratify patients and improve outcomes. In fact, to have a diagnostic and prognostic value, the biomarkers detected by IHC need to be quantified and expressed in numerical values. Different methods have been developed to quantify IHC biomarker staining for diagnostic and therapy design. So far, a universally accepted standardized unbiased method to quantify IHC does not exist. Instead, for each tumor type and correlated therapeutic regimen a specific method with a defined antibody and protocol has been developed. The most common quantification methods are combinative semi-quantitative and percentage scoring methods.1 In the percentage scoring method, the relative immune-positive cell percentage in relation to the total number of cells is evaluated and reported using numbers from 0 to 9 where each unit increase represents 10% positive staining increments. For example, if the percentage of positive cells is between 0% and 9%, the score will be 0; if it is between 10% and 19%, the score will be 1; and so on. This method is preferable in large studies to avoid batch effects and can overcome interpretation errors in the evaluation of the positive cells. A limitation is that it does not include the staining intensity which in some cases could be important for patients’ stratification.2 Combinative semi-quantitative scoring is the most used method in the current prognostic biomarker research, providing a combined positive score incorporating both quantitative and qualitative assessments. In addition to the quantitative assessment of the relative immune-positive cell percentage described above, staining intensity is also assessed. Using the combinative semi-quantitative scoring method, the intensity is usually scored from 0 to 3 with 0 indicating negative staining, 1+ weakly positive staining, 2+ moderately positive, and 3+ strongly positive staining.2 Although IHC can be evaluated both quantitatively and qualitatively, an enormous number of variations is possible. This is due to the high number of diverse staining localization and spatial combinations. This leads to divergent interpretations among pathologists and discrepancies within pre-clinical and clinical studies evidencing the need for a more uniform system to interpret and quantify IHC data. Testing for PD-L1 in metastatic NSCLC PD-L1 expression is estimated in different manners depending on the type of cancer. Interestingly, in different indications, the presence of PD-L1 is not alone indicative of therapeutic efficacy. Different clinical trials demonstrated that, for different tumor types, PD-L1 localization correlated with treatment efficacy if PD-L1 is expressed in only immune cells, only cancer cells, or both. Thus, the IHC’s ability to localize the staining is fundamental.3 As an example of IHC use in the clinic, evaluation of PD-L1 expression helps identifying metastatic NSCLC patients who are eligible for treatment with pembrolizumab. In fact, in NSCLC, PD-L1 is a proven biomarker for patient response to pembrolizumab.3,4 In general, PD-L1 staining by IHC is evaluated using a combined positive score (CPS) and tumor proportion score (TPS) depending on the cancer type. The CPS score is calculated by counting the PD-L1 positive tumor cells and mononuclear inflammatory cells, then dividing this number by the total tumor cells, and multiplying by 100.4 CPS is used in the clinic to identify candidates for treatment with pembrolizumab in indications such as gastric cancer.3 In advanced NSCLC,4 the TPS scoring method is preferred to evaluate PD-L1 expression. TPS is calculated as the number of PD-L1 positive tumor cells divided by the total number of tumor cells and then multiplied by 100. Interestingly, to determine the eligibility of a patient for pembrolizumab treatment, only PD-L1 membrane staining is scored while staining intensity is not included in the score, and only the tumor cells and not the immune cells are evaluated. In this case, a competent, trained pathologist is critical to evaluate tumor heterogeneity and discriminate between tumor cells and infiltrating macrophages that are usually positive for PD-L1. For this PD-L1 staining scoring method to be reliable, at least 100 viable tumor cells must be evaluated within the patient’s tissue.2, 4 IHC has proven to be a powerful tool for oncology clinical practice. However, in order to fully leverage the information enclosed within the tissue in a clinical setting, and given the complexity of the IHC staining interpretation, crucial are both the pathologist’s training and expertise in the specific disease evaluation. [1] Dabbs DJ. 2014. Diagnostic immunohistochemistry: theranostic and genomic applications. 4th ed. Philadelphia: Elsevier Saunders. [2] Kim S, Roh J, Park C. 2016. Immunohistochemistry for Pathologists: Protocols, Pitfalls, and Tips, J Pathol Transl Med 50(6):411-418. [4] Inamura K. 2018. Update on Immunohistochemistry for the Diagnosis of Lung Cancer, Cancers10(3), 72; [3] Davis AA, Patel VG. 2019. The role of PD-L1 expression as a predictive biomarker: an analysis of all US Food and Drug Administration (FDA) approvals of immune checkpoint inhibitors, j. immunotherapy cancer 7, 278 .
Evaluating DNA Damage in Preclinical PDX Models
DNA damage is one of the primary triggers of cancer development and has been linked to many types of cancers, including prostate, stomach, liver, and skin cancers, as well as leukemia. Within cells, the DNA sequence encodes all the instructions required for building proteins that are needed for cellular functions such as metabolism, replication, tissue, and organ maintenance. The fidelity of the DNA sequence in a cell is maintained by multiple mechanisms, including the DNA damage repair mechanism, but errors and mutations can occur, which sets off a chain of events that leads to tumor growth. DNA damage can be caused by exogenous sources, such as UV radiation, chemical carcinogens, and infection with human papillomavirus or Helicobacter pylori. Endogenous DNA damage can also be caused by multiple factors, including unchecked metabolites like reactive oxygen species (ROS) and defects in DNA damage repair enzymes. Since DNA damage mechanisms have been known to cause numerous cancers, several drugs, particularly small molecule inhibitors, have been developed to target DNA damage repair pathways. Improvements in animal models for cancer have revolutionized how anti-cancer drugs are evaluated and developed, including drugs targeting DNA damage. Patient-derived xenograft (PDX) models have been particularly powerful tools since they use patient-derived tumor tissue engrafted into mice. Tumor cell lines, solid tumor tissue, or hematological tumors can be transplanted into immunodeficient mice to study DNA damage repair mechanisms. These immunocompromised mice can also be humanized by inoculating human immune cells or made to express components of the human immune system, like immune checkpoint molecules, to better screen for the effectiveness of various anti-cancer treatments. In this era of rapid, high-throughput DNA sequencing, individual tumors can be sequenced and specific defects in DNA damage repair pathways can be defined. This same tumor tissue can be engrafted into a PDX mouse model for screening of drugs or therapeutics that tackle the appropriate DNA damage repair defect. Different assays can also be used to study DNA damage and repair mechanisms: comet assay is used to detect DNA single/double-strand breaks in single cells, γH2AX detection by IHC staining or western blot can be used to detect, measure, and localize DNA breaks. This approach is powerful for screening preclinical drug candidates targeting DNA damage and repair for efficacy against a range of tumors and it also provides insights into potential off-target effects or toxicities. From the patient's perspective, pre-screening potential treatment options in mice can lead to the selection of the most appropriate drug or therapeutic targeting DNA damage and repair and help avoid treatments that may be ineffective. DNA damage events can lead to tumor growth and this area of research continues to inform drug development on the bench and patient care in the clinic. Champions Oncology is the ideal partner to accelerate your DNA damage-targeted drug pipeline. We offer a large bank of PDX models, particularly PDX models with high microsatellite instability (MSI-H), that have dysregulated DNA damage repair mechanisms, for ex vivo and in vivo studies. We also provide DNA sequencing, comet assay, and γH2AX detection by IHC staining or western blot, to support your DNA damage targeting drug programs. To add DNA damage evaluation to your upcoming study, contact us today.

Blotting Basics - Western Blot Applications in Preclinical Oncology Research
Western blotting is a decades-old laboratory technique that is used to detect specific proteins from cell culture, tissue, or blood specimen. The term “western blot” is a twist on the Southern blotting method developed by Edwin Southern, which is used to detect DNA and shares methodological similarities with western blotting. The western blot method was first described by Harry Towbin in 1979 but the term “western blot” was coined by W. Neal Burnette in 1981[1],[2]. Since its initial description, the application of western blotting to all fields of biological and biomedical research has been broad because it is a straightforward and robust method for detecting specific proteins. Here we provide an overview of the western blot method and highlight the current application of western blotting in preclinical oncology research. Western Blot Basics The western blot technique can be used to separate and identify a specific protein using three major steps: 1. Size separation using gel electrophoresis, 2. Transfer to a solid membrane, and 3. Detection with a specific antibody. Most western blot methods begin with a lysate of cells or tissue, which releases a mix of proteins that are separated by molecular weight using gel electrophoresis. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) is the most common type of gel electrophoresis for western blotting and includes a protein denaturation step prior to gel electrophoresis such that proteins are separated by molecular weight. The SDS buffer causes proteins to become negatively charged so electrophoresis allows for migration of proteins from smallest to largest weight towards a positive charge. After gel electrophoresis, proteins are transferred to a solid membrane, usually polyvinylidene difluoride or nitrocellulose, using electroblotting or a slower alternative method based on capillary action. This membrane can now be probed with a primary antibody specific to a protein of interest and the primary antibody is visualized using a secondary antibody that recognizes a species-specific region of the primary antibody and is conjugated with a chemiluminescence substrate for visualization. Other less common visualization methods use colorimetric substrates or radioactive labels on secondary antibodies. Application of Western Blotting to Preclinical Studies Why is western blotting a method that is still used after more than forty years since its development? Western blotting remains a reliable, affordable, and practical method for detecting specific proteins. The application of western blotting in preclinical oncology research allows to validate high throughput single-cell RNA sequencing or proteomics methods that detect elevated proteins associated with specific cancers[3]. Western blotting can also validate tissue microarray and immunohistochemistry findings with respect to specific proteins that are overexpressed in tumor tissue. Together these data can be used toward developing prognostic biomarkers for cancers, such as measuring overexpression of Kin of IRRE-like Protein 1 (KIRREL) in breast cancer and precancerous tissue[4]. Western blotting is also a useful tool for understanding molecular mechanisms that drive cancer progression by measuring expression of critical proteins such as HMGB1, cyclins, and various oncogenes[5],[6]. Western blot has made a leap into the twenty-first century with advancements in process workflow and sensitivity using new platforms like ProteinSimple’s JESS System. Western blot will continue to be a workhorse for detecting and monitoring specific protein expression and the application of western blotting continues to be broad in preclinical and clinical oncology studies related to understanding oncogenesis and defining potential tumor biomarkers.
.jpg)
Using Flow Cytometry as an In Vivo Study Endpoint
In vivo models for numerous diseases and conditions have endpoints that have involved animals being gravely ill or dying. As researchers have sought to utilize animal models in more humane and practical ways, surrogate endpoints have been developed that prevent animals from suffering and provide critical research data. Flow cytometry has been instrumental to these advances. Consider these aspects of preclinical flow cytometry endpoint analysis as you develop new protocols. 1. What are the immune system features of your disease state? Flow cytometry provides the most useful data when the cell subsets of interest are well-defined and robust. You may need to analyze existing research literature or do pilot studies to define the immune cell subsets of interest for a particular disease state, be it changes in regulatory T cells in the tumor microenvironment, or the proliferation of plasmablasts in different leukemias. You must identify which profound changes in different cell populations are most closely correlated with morbidity and mortality in your animal model. 2. What is the desired treatment outcome? Preclinical studies with surrogate endpoints are valuable for screening potential therapeutic candidates. These drugs or biologics may have undesirable off target effects as well. In designing a flow cytometry assay for alternative endpoints, it is critical to identify the changes in immune cell subsets that reflect therapeutic improvements or indicate potential toxicity or off target effects. 3. Can this be translated into a clinical flow cytometry protocol? In some disease models, particularly models using humanized mice, flow cytometry endpoints can be used in both preclinical screens and to evaluate clinical trial specimens. This consideration is valuable as protocols are developed and cell phenotypes are identified as predictors of good or poor prognoses. Flow cytometry endpoint analysis not only advances the humane use of animal models but can be translated into informative clinical protocols that are critical for the evaluation of potential therapies.

Factors to Consider When Selecting Next-Generation Sequencing (NGS) Technology
Next-generation sequencing (NGS) technology has transformed the biomedical research landscape. Only a few years ago, high resolution genome or exome sequencing would be cumbersome and cost-restrictive, but current NGS technology platforms now allow for basic and clinical researchers to include these approaches for routine DNA and RNA sequencing needs. What are the different NGS sequencing approaches and how are they applied to oncology research? 1. Whole Genome Sequencing (WGS): NGS technology can be used for WGS of human genomes and tumor-specific genomes, as well as animal model and microbial genomes. WGS produces high resolution genomic sequences of expressed genomic regions as well as unexpressed regulatory regions. For preclinical oncology research, WGS is critical for characterizing genomic profiles associated with tumor progression or potential responsiveness to targeted drug therapies. WGS can detect single nucleotide variants, copy number variants, and insertions/deletions in tumor cells[1]. The comprehensive scope of WGS makes it well suited for detecting mutations in both coding and non-coding regions[2]. WGS is also useful for population level oncology studies that evaluate genetic susceptibility to specific cancers and potential heritability[3]. 2. Whole Exome Sequencing (WES): WES techniques focus on sequencing the exome, which are comprised of protein expressing regions, or exons, within the genome. WES is an appropriate method for identifying genetic mutations that alter protein sequences, and WES data can be used toward measuring the tumor mutational burden (TMB) and predicting treatment efficacy[4]. WES data can also be used to identify potential new drug targets or mechanisms of drug resistance[5]. 3. Targeted Sequencing: This method focuses on defined gene regions and is typically used in diagnostic applications or for validation of WGS or WES results. Targeted sequencing works well for screening tumor samples for well characterized mutations, such as those associated with BCL2, BRCA-1/2, BRAF, and EGFR, and can be used for identifying appropriate targeted therapies[6]. 4. RNA sequencing: RNA sequencing is now emerging as a powerful tool that complements NGS DNA methods because the transcriptional profile of a single cell can be measured and used to bridge genomic data with cellular phenotypes. Single-cell RNA sequencing (scRNA-seq) has specifically emerged as a powerful method for understanding the heterogeneity of cell populations within a tumor. Together with histological data, scRNA-seq data can be used to distinguish between neoplastic cells, immune cells, and healthy cells from the surrounding tissue, and it can also be used to evaluate how experimental treatments alter the tumor microenvironment[7]. NGS methods are transforming both basic oncology research and clinical care, from identifying novel mutations to pinpointing personalized cancer therapies. Each method is suited to specific applications, so working with experts in NGS technology is critical to method selection and data analysis. 1 Bewicke-Copley F et al. Applications and Analysis of Targeted Genomic Sequencing in Cancer Studies. Comput. Struct. Biotechnol. 2019;17: 1348-1359. 2 Nakagawa H, Fujita M. Whole Genome Sequencing Analysis for Cancer Genomics and Precision Medicine. Cancer Sci. 2018;109(3):513-522. 3 Rotunno M et al. A Systematic Literature Review of Whole Exome and Genome Sequencing Population Studies of Genetic Susceptibility to Cancer. Cancer Epidemiology and Prevention Biomarkers. 2020;29(8):1519-34. 4 Klempner SJ et al. Tumor Mutational Burden as a Predictive Biomarker for Response to Immune Checkpoint Inhibitors: A Review of Current Evidence. Oncologist. 2020 Jan;25(1): e147-e159. 5 Beltran H et al. Whole-Exome Sequencing of Metastatic Cancer and Biomarkers of Treatment Response. JAMA Oncol. 2015;1(4):466-474. 6 Vestergaard LK et al. Next Generation Sequencing Technology in the Clinic and Its Challenges. Cancers (Basel). 2021;13(8):1751. 7 Fan J et al. Single-Cell Transcriptomics in Cancer: Computational Challenges and Opportunities. Exp Mol Med. 2020; (52)1452–1465.