Chronic lymphocytic leukemia (CLL) is one of the most common forms of adult leukemia, and its chronic nature has made it a challenging blood cancer to completely cure. CLL affects B cells and is typically classified into two categories: little or no somatic hypermutation in the immunoglobulin heavy chain variable region (IGHV), called unmutated CLL, or high mutation levels in the IGHV gene, called mutated CLL.[1] Unmutated CLL is more aggressive than mutated CLL, and the presence of these abnormal IGHV sequences, a part of B cell receptors (BCR), leads to abnormal BCR signaling and the uncontrolled proliferation of leukemic cells.
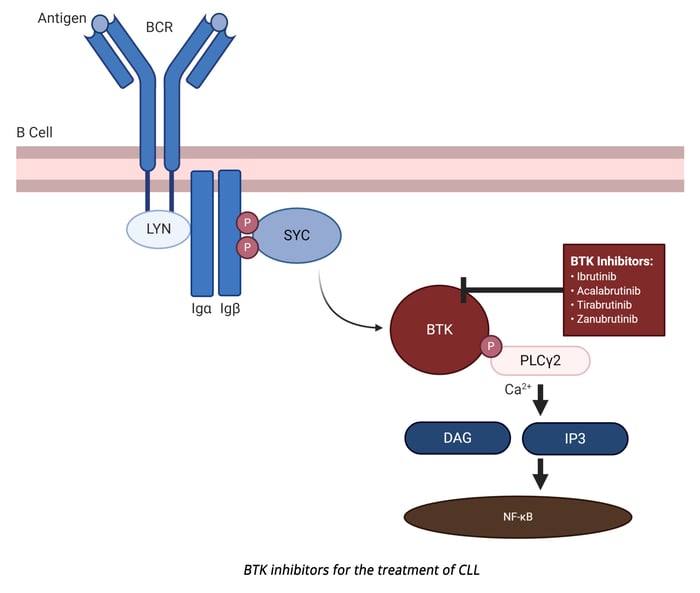
Bruton’s tyrosine kinase (BTK) is an essential enzyme downstream from the BCR and is responsible for propagating the signaling cascade initiated by BCR engagement with pathogen-associated antigens.[2] Notably, BTK has been shown to be constitutively active in CLL and drives both leukemic cell proliferation and lymph node homing.[3] As antigen binds to the BCR, multiple protein tyrosine kinases are activated via interactions with the cytoplasmic domain of the BCR, including Lyn and Syk, as well as translocation of BTK to the plasma membrane through interactions with phosphatidylinositol-3,4,5 (PIP3).[4] Lyn and Syk phosphorylate BTK to activate multiple non-receptor protein tyrosine kinase signaling pathways, including NF-κB, MAPK, and phospholipase C gamma (PLCγ). Under normal conditions, this pathway leads to controlled B cell proliferation and differentiation, however, this same pathway leads to uncontrolled B cell proliferation in malignancies, such as CLL.
The role of BTK in CLL has drawn researchers to develop BTK inhibitors, such as ibrutinib - an FDA (Food and Drug Administration)[5] and EMA (European Medicines Agency)[6] - approved CLL treatment. Ibrutinib forms a covalent bond with a cysteine residue (C481) at BTK’s active site, and thus inhibits kinase activity, including autoactivation.[3] This kinase inhibition shuts down BCR signaling, reduces B cell proliferation, and promotes apoptosis of leukemic cells. Ibrutinib is effective as a standalone treatment for mutated and unmutated CLL and when combined with rituximab is an effective therapy for relapsed CLL.[1] In general, ibrutinib is well tolerated and shows continued efficacy during extended treatment periods.[7]
While ibrutinib has been a clinical success, there are a subset of CLL patients who develop resistance to this therapeutic. This resistance has been mapped to a C481S mutation that prevents ibrutinib from covalently binding to BTK resulting in continuous BCR signaling within leukemic cells.[3] Next-generation, reversible BTK inhibitors, such as ARQ 531, are currently being developed for relapsed/refractory CLL. ARQ-531 is a non-selective BTK inhibitor that suppresses signaling in cells with C481S BTK and PLCγ mutations. ARQ-531 also has an additional inhibitory activity against ERK signaling.[8] Furthermore, the FDA has recently approved LOXO-305, a non-covalent, reversible BTK inhibitor. LOXO-305 inhibits signaling in cells with wild-type or C481S-mutated BTK.[9, 10] In contrast, acalabrutinib, a second-generation irreversible BTK inhibitor has also been approved by the FDA. Acalabrutinib has a shorter half-life, allowing for variable dosages, and has increased C481 specificity that enhances its BTK inhibitory effects. The biochemical properties of acalabrutinib make it a suitable option for patients with treatment naïve CLL.[11]
In January 2023, the FDA and EMA approved an additional second-generation irreversible BTK inhibitor, zanubrutinib, for CLL.[12] Zanubrutinib’s design was guided by a structure-activity strategy to generate sustained BTK occupancy. Zanubrutinib exhibits reduced ITK and EGFR inhibition and has 4x longer half-life than acalabrutinib.[13] As such, it persists at high concentrations within the body making it available to re-inhibit newly synthesized BTK proteins, a unique difference from ibrutinib and acalabrutinib.[14]
In a phase I/II study, all CLL patients treated with zanubrutinib had complete and sustained BTK occupancy in peripheral blood mononuclear cells and lymph nodes.[15] Zanubrutinib’s performance supports the hypothesis that increased BTK selectivity maximizes BTK inhibition and drug efficacy. Additionally, zanubrutinib’s enhanced BTK specificity reduces the incidence of off-target toxicities which may reduce side effects commonly associated with ibrutinib, such as cardiac events, subdural hematomas, and gastrointestinal bleeding.[10, 16, 17] This is particularly relevant to CLL as this class of BTK inhibitors are used by CLL patients indefinitely.
BTK inhibitors have become an invaluable CLL treatment and will continue to improve and provide therapeutic benefits. Additional studies such as NCT03734016 are underway to evaluate the effects of zanubrutinib and BTK inhibitors with other targeted therapies, which may improve the efficacy and durability of these treatments.