
Trends in Oncology
A KRAS mutation is not a strategy: Why allele, co-mutation, and tumor type matter
In our last blog post, we explored why resistance has become the defining challenge in KRAS drug development. As the field has matured, it has become clear that early activity alone is not enough, and understanding why tumors respond or not, and how they escape treatment is now essential. It also raises the deeper issue of if response and resistance vary so widely, then a critical assumption at the heart of many programs needs to be challenged: Can “KRAS-mutant” still be treated as a single category in drug development? The evidence increasingly suggests the answer is no. The problem with treating KRAS as one market As KRAS-targeted therapies have moved forward, many development strategies have been framed broadly around “KRAS-mutant tumors.” On the surface, this makes sense as KRAS is one of the most common oncogenic drivers across multiple cancer types. However, in practice, this simplification hides important biological differences. Across KRAS-mutant tumors, responses to targeted therapies are highly variable, with some settings showing meaningful activity, while others demonstrating limited or short-lived responses. Even within the same tumor type, outcomes can differ significantly across patients. This variability is not random and reflects the underlying biological context, which now matters more than ever. Allele matters more than ever Not all KRAS mutations are the same, with variants such as G12C, G12D, and G12V differing in their biochemical properties, downstream signaling behavior, and sensitivity to targeted inhibition. As more therapies are developed beyond the initial wave of KRAS G12C inhibitors, these differences are becoming increasingly relevant. Emerging preclinical data shows that response patterns can vary across KRAS allele types, influencing both the depth and durability of response. For drug developers, this means that evaluating an asset against a generic “KRAS-mutant” panel is no longer sufficient, and the real question to now ask is: Which KRAS variants does your therapy work against, and under what conditions? Co-mutations shape response and resistance KRAS does not operate in isolation. Tumors often carry additional genetic alterations that influence pathway signaling, tumor behavior, and therapeutic response. These co-driver mutations can enhance sensitivity in some contexts or drive resistance in others. Across clinically relevant models, differences in co-mutation profiles have been linked to distinct response patterns and resistance mechanisms, which has two important implications: A therapy that appears broadly effective in a simplified model system may fail in more complex, clinically representative settings. Co-mutation context can represent an opportunity. Understanding these relationships can help identify patient subsets that are more likely to benefit or reveal combination strategies that overcome resistance. Tumor type defines the biological landscape KRAS mutations occur across multiple tumor types, including non-small cell lung cancer, colorectal cancer, and pancreatic ductal adenocarcinoma. Each tumor type exists within its own biological environment, shaped by factors such as tissue origin, microenvironment, and lineage-specific signaling networks. These differences influence how KRAS-driven pathways behave and how tumors respond to targeted therapies. As a result, a therapy that performs well in one indication may not translate directly to another. This is why preclinical strategies must be designed with indication in mind. Testing a KRAS program in the wrong context can lead to misleading conclusions about its potential. Why context-aware model selection is critical Taken together, these factors make one thing clear: Context is a requirement, rather than a refinement. Effective KRAS studies must move beyond generic model selection and instead reflect the biological diversity seen in the clinic, which includes: Selecting models that represent specific KRAS variants Capturing diverse co-mutation landscapes Matching studies to relevant tumor types Incorporating clinically meaningful patient characteristics Modern preclinical platforms are increasingly designed to meet these needs by combining deeply characterized patient-derived models with molecular and clinical annotation. This approach allows teams to design studies that are not only more realistic, but also more informative. Better context leads to better decisions When KRAS programs are evaluated within the right biological context, the quality of decision-making improves significantly, allowing teams to: Rank assets more accurately Identify the most promising indications Develop more precise biomarker strategies Design more rational clinical trials Perhaps, most importantly, they can avoid overinterpreting results that may not translate outside of a narrow experimental setting. In a competitive and rapidly evolving field, this clarity can make the difference between advancing the right candidate and pursuing a path that ultimately fails in the clinic. From resistance to real-world biology In our previous article, we highlighted the importance of modeling resistance to understand how tumors respond over time, and in this post we have expanded on that idea. Resistance does not emerge in a vacuum. It is shaped by the biological context of the tumor, including allele, co-mutation, and tumor type. Ignoring that context risks misunderstanding both response and resistance. What comes next If biological context shapes response, then treatment history is another critical factor that must be considered. In reality, most patients entering clinical trials are not treatment-naive. Their tumors have been shaped by prior therapies, and those therapies influence how they respond to potential new agents. In our next post in this series, we will explore why treatment-naive models are increasingly insufficient and why pretreated, clinically aligned models are essential for studying KRAS programs in today’s landscape. Talk to us If you are developing a KRAS-targeted therapy, understanding how allele, co-mutation, and tumor type shape response is no longer optional. Talk to our team about designing context-aware preclinical studies that generate data you can trust and act on. Have a Question?

The next KRAS battleground: modelling resistance before the clinic
In our previous posts, we explored how KRAS has entered a new era and why benchmarking has become essential for differentiating assets in an increasingly competitive landscape. But as the field evolves, a new reality is becoming clear: In KRAS drug development, early activity is no longer enough. The programs that succeed will be the ones that understand resistance before they reach the clinic. The shift from proof of concept to durability For decades, KRAS was defined by a single challenge: proving that it could be drugged at all, a barrier that has now been crossed. With multiple KRAS and pan-RAS targeted therapies advancing through the pipeline, the question is no longer whether a compound can generate tumor shrinkage, it is whether that response will be deep, durable, and reproducible across patient populations, and is where many programs begin to diverge. Preclinical studies often still focus on early efficacy signals and in reality, clinical success depends on something much more complex: how tumors respond over time and how quickly they adapt. Why resistance is now the defining challenge in KRAS Recent preclinical work in KRAS-mutant models shows that responses to targeted therapies are highly variable, with clear differences in both intensity and duration. Some tumors fail to respond at all, with others responding initially and then relapsing as resistant clones emerge. Understanding these patterns is now critical to making informed development decisions. Across diverse KRAS-mutant settings, response heterogeneity has been linked to KRAS allele type, co-driver mutations and prior treatment exposure. These variables reflect the biological diversity that defines real patient populations and ignoring them in preclinical work means overlooking the very factors that determine clinical outcomes. Not all resistance is the same To model resistance effectively, it is important to recognize that there are two distinct challenges: 1. Intrinsic resistance: Some tumors show little or no response to treatment from the outset. These cases often reflect underlying biology that makes the therapy ineffective in that context. 2. Acquired resistance: Other tumors respond initially, but over time develop mechanisms that allow them to escape treatment pressure. Advanced KRAS studies now capture both dynamics by: Comparing responders versus non-responders at baseline Tracking tumors longitudinally after initial regression Analyzing the molecular changes that drive resistance over time Without this dual perspective, programs risk misunderstanding both the limits and the true potential of their assets. Why traditional preclinical models fall short So, if you know that resistance is the defining challenge, why do so many preclinical studies fail to capture it? The answer usually lies in how those studies are designed. Many studies designs still rely on small numbers of models, treatment-naive systems and single endpoint efficacy readouts. These approaches can identify whether an asset has activity. They struggle to answer more important questions, like: Which patients are likely to respond? How durable are those responses? What mechanisms will drive relapse? Clinically relevant preclinical platforms are moving toward combining diverse patient-derived models, treatment history, and deep molecular characterization to better reflect real-world tumor biology. This shift now makes it possible to evaluate how therapies perform beyond the initial response. What traditional KRAS studies miss (side by side comparison) Traditional studies Resistance-aware studies Few models Diverse cohorts Treatment-naive Pretreated Single endpoint Longitudinal Limited readouts Multi-omic What better KRAS studies should capture It is clear, to move from proof of concept to true translational insight, KRAS studies need to evolve. At a minimum, that means incorporating: Biological diversity across KRAS variants and tumor types Co-mutation context to reflect real genetic drivers Treatment history to model clinically relevant resistance Longitudinal sampling to track response and relapse Multi-omic profiling to uncover mechanisms of sensitivity and escape These elements allow teams to move beyond simple efficacy metrics and begin to answer the questions that matter most for clinical success. Why resistance modeling changes decision making When resistance is built into preclinical strategy, the impact directly informs things like; Asset ranking between competing KRAS programs, Indication selection across tumor types; Biomarker strategy for patient segmentation, and; Combination approaches designed to prevent or overcome resistance. Resistance modeling is not just about understanding failure. It is about designing for success earlier in development. From benchmarking to biology Our previous post highlighted why benchmarking is now essential in KRAS drug development. However, benchmarking alone is not enough. To generate meaningful comparisons, studies must account for the biological factors that drive response and resistance. Without that context, even rigorous comparisons can lead to misleading conclusions. What comes next If resistance varies so widely across KRAS-mutant tumors, it raises an important question: Can “KRAS-mutant” still be treated as a single category in drug development? In our next blog post, we explore why the answer is no and why allele, co-mutation, and tumor type must shape every aspect of KRAS strategy. If you are advancing a KRAS program, designing studies that capture both response and resistance is no longer optional. Talk to our team about building preclinical strategies that reflect real patient biology and generate data you can act on. Have a Question?

Why Benchmarking is Now Essential in KRAS Drug Development
The KRAS landscape has changed. As next-generation inhibitors continue to demonstrate clinical impact, the expectations placed on preclinical programmes are increasing. At the centre of this shift is a simple but critical concept. Benchmarking is no longer a differentiator, but a requirement. The Rise of a Clinical Reference Point As therapies such as daraxonrasib advance through clinical development, they establish a new baseline for efficacy. This baseline becomes the reference against which all new KRAS-targeting programmes are evaluated. For preclinical scientists, this fundamentally changes the design of studies. It is no longer sufficient to show that a compound reduces tumor growth in a subset of models. The real question is whether that activity represents meaningful improvement over what is already achievable. Without benchmarking, this question cannot be answered. What Benchmarking Enables Benchmarking provides context and transforms isolated, siloed data into translatable data; the basis on which your decision-making can be trusted. When preclinical results are generated against a known standard, researchers can: Quantify relative efficacy Identify tumor subtypes where a therapy may outperform existing options Understand where resistance patterns differ Evaluate the potential for combination strategies In KRAS-mutant tumors, where heterogeneity is high and resistance is common, this context is essential for making informed decisions. The Importance of Scale and Annotation Not all benchmarking datasets are equal. To be meaningful, benchmarking must be built on three core components: 1. A sufficiently large and diverse model set: KRAS mutations span multiple tumor types and molecular contexts, and a robust dataset must reflect this diversity. 2. Deep molecular characterisation: Genomics and transcriptomics alone are not enough to fully capture tumor biology. Protein-level and pathway-level data provide additional resolution that is critical for understanding response. 3. Clinical relevance: Models must reflect real patient biology, including treatment history and resistance mechanisms. At Champions Oncology, we have generated daraxonrasib response data across more than 50 KRAS-mutant PDX models spanning lung, colorectal, and pancreatic cancers. Each model is fully annotated with clinical and molecular data, enabling direct linkage between response and biology. This is not a conventional screening dataset but a translational framework enabling decision-making. Moving Beyond Binary Outcomes Traditional preclinical studies often present results in binary terms. Tumors respond or they do not. While this can be useful at an early stage, it lacks the resolution needed for modern drug development. Benchmarking enables a more nuanced view and instead of asking whether a tumor responds, scientists can ask: How does this response compare to existing therapies? What molecular features distinguish responders from non-responders? Which resistance pathways are likely to emerge? This level of analysis is essential for designing therapies that succeed in the clinic. Implications for Clinical Strategy The value of benchmarking extends beyond the preclinical stage and directly informs clinical development. By linking response data to molecular features, benchmarking datasets can support: Biomarker-driven patient selection Rational combination therapy design Selection of optimal indications Identification of expansion cohorts In a competitive KRAS landscape, these insights provide a critical advantage. A New Standard for Preclinical Research As the field continues to evolve, benchmarking will become a standard component of preclinical workflows. Teams that fail to incorporate benchmarking risk generating data that lacks context and relevance. The future of KRAS research lies in integrated, data-rich approaches that connect biology, pharmacology, and clinical strategy. Benchmarking is the foundation of that future. What Comes Next In the next blog in this series, we examine why many KRAS preclinical models fail to translate into clinical success and how integrated, multi-omic approaches are addressing this gap. Continue exploring KRAS blog posts

KRAS Has Entered a New Era. Why Your Preclinical Strategy Must Change Now
KRAS has long been one of the most important targets in oncology, and for decades it was widely considered “undruggable”. Fortunately, that perception has changed rapidly and today, KRAS-targeted therapies are no longer theoretical. They are clinically validated, increasingly effective, and reshaping how drug development programmes are designed. The transition from early KRAS inhibitors (KRASi) to next-generation compounds represents a fundamental shift in how our field approaches targeted therapy. For preclinical and translational teams, this shift introduces both opportunity and risk. Programs that adapt quickly will be well positioned to succeed, whereas those that rely on outdated models and assumptions may fall behind. A New Standard of Care Is Emerging Recent clinical data for pan-RAS(ON) inhibitors, such as daraxonrasib, signal a turning point in KRAS drug development. As these therapies move toward becoming standard of care, they establish a new benchmark against which all future KRAS-directed agents will be measured. This has immediate implications for preclinical strategies, and it is no longer sufficient to demonstrate activity in isolation. Every new therapy must now be evaluated relative to an evolving clinical reference point. For companies developing therapies in KRAS-mutant indications such as non-small cell lung cancer (NSCLC), colorectal cancer, and pancreatic cancer, the key question has changed. It is no longer “Does this work?” but “Does this outperform or complement what already exists?”. Why Traditional Preclinical Approaches Are No Longer Enough Historically, KRAS drug development has relied heavily on simplified systems such as cell lines or limited in vivo studies. While these approaches played an important role in early discovery, they are insufficient for today’s demands. KRAS biology is highly context dependent and mutation status alone does not determine response. Co-occurring genomic alterations, transcriptional programmes, and protein-level signalling all contribute to therapeutic sensitivity and resistance. As a result, preclinical systems that lack clinical relevance or molecular depth fail to capture the complexity of real tumours. This creates a disconnect between preclinical findings and clinical outcomes, increasing the risk of late-stage failure. The Role of Clinically Relevant Models To address this challenge, there is a growing shift toward clinically relevant models such as patient-derived xenografts (PDXs) with integrated molecular characterisation. These models enable a deeper understanding of tumor biology and provide a more accurate representation of patient response. Importantly, when these models are combined with longitudinal data and treatment history, they allow researchers to study both intrinsic sensitivity and acquired resistance. This is essential in KRAS, where resistance is not an exception but an expected outcome. Benchmarking as the New Foundation In this evolving landscape, benchmarking is becoming the basis of an effective preclinical strategy, and access to datasets that include response to current or emerging standards of care allow scientists to: Identify responder and non-responder populations Understand the molecular drivers of response Design rational combination strategies Prioritise assets with the highest likelihood of success At Champions Oncology, we recognised that benchmarking would become essential in the KRAS space. Our work has focused on building clinically annotated datasets that connect tumor response directly to molecular biology. This enables a level of insight that extends far beyond traditional screening approaches. From Activity to Decision-Making The ultimate goal of preclinical research is not only to generate data, but to inform your decision making. In KRAS drug development, the stakes are high. Clinical trials are expensive, timelines are long, and the competitive landscape is rapidly evolving. Preclinical data must therefore do more than demonstrate activity. It must guide: Patient selection strategies Combination therapy design Clinical trial structure Pipeline prioritisation This requires a shift from descriptive models to more predictive, translational systems. Looking Ahead KRAS has entered a new era. The science is advancing, the clinical landscape is evolving, and expectations are rising across the industry. For preclinical teams, this is a moment to reassess how studies are designed, how models are selected, and how data are interpreted. The organisations that lead in this space will be those that align their strategy with the realities of the current landscape, not the assumptions of the past. In the next blog in this series, we explore why benchmarking is no longer optional in KRAS research and how it is reshaping preclinical decision-making. Poster: Molecular determinants of sensitivity and resistance to the pan-RAS(ON) inhibitor daraxonrasib
Daraxonrasib Is Rewriting the Standard of Care. Here's Why Preclinical Benchmarking Matters Now
A Standing Ovation and a New Standard of Care At the 2026 ASCO Annual Meeting, the RASolute 302 Phase 3 trial delivered what many are calling the most significant advance in pancreatic cancer treatment in decades. Daraxonrasib, a first-in-class oral RAS(ON) multi-selective inhibitor developed by Revolution Medicines, nearly doubled overall survival (OS) in previously treated metastatic pancreatic ductal adenocarcinoma (PDAC), achieving a median OS of 13.2 months compared to 6.7 months with standard chemotherapy (HR 0.40; p<0.0001). Progression-free survival followed the same trajectory: 7.3 months versus 3.5 months. The results, published simultaneously in The New England Journal of Medicine, drew a standing ovation during the plenary session. ASCO's chief medical officer called it "a grand slam," and the ASCO-selected commentator described starting to cry in clinic after seeing the data. This is no longer a drug to watch. Daraxonrasib is poised to become the new standard of care for second-line metastatic PDAC, with first-line combination trials already underway. For drug developers working in KRAS-driven cancers, the question is no longer whether daraxonrasib will reshape the treatment landscape, but how fast, and whether your preclinical program is ready. Why Benchmarking Against Daraxonrasib Is Now Essential More than 90% of pancreatic cancers harbor KRAS mutations, making it one of the most RAS-addicted tumor types in oncology. As daraxonrasib transitions from investigational therapy to standard of care, it will become the benchmark against which new agents, novel combinations, and next-generation therapies are measured. This shift has immediate implications for preclinical strategy. Scientists developing therapies in KRAS-mutant NSCLC, colorectal cancer, and PDAC need access to clinically annotated models with existing daraxonrasib response data to design meaningful benchmarking and combination studies. Without this data, preclinical programs risk testing against an outdated treatment landscape. Yet despite the urgency, a significant gap exists. Most contract research organizations and preclinical providers do not have daraxonrasib response data, the KRAS-mutant patient-derived xenograft (PDX) models needed to generate it, or the multi-omic depth required to interpret results in a translational context. Champions Oncology Has Already Built the Dataset At Champions Oncology, we recognized early that daraxonrasib would become a defining compound in the KRAS space. That is why we have already screened over 50 KRAS-mutant PDX models across NSCLC, colorectal cancer, and PDAC, generating a unique and comprehensive daraxonrasib benchmarking dataset that is available today. Figure 1: Tumor response and genomic landscape of KRAS-mutant PDX models treated with Daraxonrasib (RMC-6236) This is not a standard drug screen. Every model in the cohort is fully annotated with clinical history, treatment status, and deep molecular profiling, including whole exome sequencing, RNA sequencing, and integrated genomic analysis. The resulting waterfall plots and genomic annotation panels allow scientists to rapidly identify responder and non-responder models matched to their therapeutic hypothesis, and to understand the molecular context driving each outcome. Understanding what drives resistance. Differential gene expression analysis of responding versus non-responding KRAS G12D-mutant PDAC models has identified transcriptional programs associated with primary resistance to daraxonrasib, providing actionable insights for biomarker development and patient selection strategies. Modeling acquired resistance in real time. Because secondary resistance is an inevitable feature of targeted therapy, we have developed acquired resistance models under continuous daraxonrasib treatment pressure. Models CTG-2473 (pancreatic, KRAS G12D) and CTG-0068 (colorectal, KRAS G12D) both demonstrated initial tumor regression followed by regrowth, recapitulating the resistance patterns expected in patients. These models create a powerful translational framework for testing next-line agents, rational combination strategies, and treatment sequencing approaches. Going deeper with multi-omic prediction. Champions' Pharmaco-Pheno-Multiomic (PPMO) integration platform takes this further by layering whole-cell proteomics, cell surface proteomics, genomics, and transcriptomics across 56 KRAS-mutant PDX models treated with daraxonrasib. The resulting computational model achieves 80% prediction accuracy for drug response, substantially outperforming RNA-only approaches, which plateau at approximately 60%. This level of biological resolution is critical for identifying the molecular determinants of sensitivity and resistance that standard biomarker panels miss. Download the Full Poster These findings were first presented at AACR 2026 (Poster 1884): Molecular Determinants of Sensitivity and Resistance to the Pan-RAS(ON) Inhibitor Daraxonrasib (RMC-6236) Across KRAS-Mutant Patient-Derived Models. For the complete dataset, including response profiles, genomic annotations, and resistance modeling data, download the full poster here. For additional context, read our companion blog: Understanding Sensitivity and Resistance to Pan-RAS(ON) Inhibition Across KRAS-Mutant Tumors. The Landscape Is Shifting. Let's Talk About Your Program. Daraxonrasib is changing how KRAS-targeted therapies will be developed and evaluated. Whether you are benchmarking a novel compound against an emerging standard of care, designing combination strategies, or investigating mechanisms of primary and acquired resistance, we have the models, the data, and the translational depth to support your program. Contact us today to discuss your study
Scaling Chem-Seq and Functional-Seq to Decode Drug and Genetic Perturbations in Patient-Derived Organoids
Large-scale drug discovery increasingly depends on understanding mechanism, not just measuring viability. While high-throughput screening has traditionally relied on simple 2D systems, these models often fail to capture the biological complexity of human tumors or provide the mechanistic depth needed to make confident development decisions. At the same time, transcriptomic approaches have historically been too low-throughput to support large compound libraries. In our AACR 2026 poster, we introduced scalable Chem-Seq and Functional-Seq platforms designed to close this gap. By combining high-throughput transcriptomic profiling with patient-derived organoid models, we can systematically map chemical and genetic perturbations at scale while preserving clinically relevant tumor biology. Why transcriptomic profiling matters in drug discovery Phenotypic screening alone rarely explains why a compound works or fails. Two agents may produce similar viability effects for completely different reasons, while others may appear inactive despite engaging their intended targets. Transcriptomic profiling addresses this challenge by capturing pathway-level responses and signatures of mechanism of action. However, traditional RNA-seq workflows are difficult to scale. Sample preparation, library generation, and sequencing cost all become limiting factors. Our goal was to develop a workflow that preserves transcriptomic resolution while enabling tens of thousands of perturbations per week in relevant cancer models. A scalable Chem-Seq workflow built for throughput Our Chem-Seq platform is built around DRUG-seq™, a workflow optimized for bulk mRNA profiling directly from cancer cell lines or PDX-derived organoids in 384-well plates. Cells or organoids are plated, cultured, and treated with small-molecule compounds. Following treatment, a single-step lysis and barcoded oligo-dT capture allows mRNA from each well to be uniquely indexed in situ. After reverse transcription, all wells are pooled, amplified, and processed through standard Illumina library preparation workflows. Libraries are sequenced to a depth sufficient to capture robust transcriptional signatures while maintaining scalability. This approach supports roughly 50,000 perturbations per week, spanning small molecules, and siRNAs. The key advantage is efficiency. By eliminating many traditional bottlenecks, Chem-Seq enables rapid, reproducible transcriptomic profiling at a scale that aligns with modern screening demands. From raw reads to mechanism-resolved insights Chem-Seq data are processed to generate gene-by-barcode matrices, followed by normalization, batch correction, and dimensionality reduction. Principal component analysis and UMAP visualizations enable rapid triage of perturbation effects, while differential expression and pathway analyses reveal mechanism-associated signatures. To further contextualize results, Chem-Seq signatures are compared against large public perturbation datasets using connectivity mapping approaches. This allows unknown compounds to be matched to known drugs, siRNA knockdowns, or CRISPR perturbations based on shared transcriptional responses, providing early insight into mechanism of action and potential off-target effects. Anchoring chemistry with Functional-Seq While chemical perturbations provide rich information, interpreting their biological meaning is often strengthened by genetic context. Functional-Seq integrates siRNA-mediated knockdown of drug targets directly into the same transcriptomic framework. In this study, Functional-Seq was applied in non-small cell lung cancer PDX organoid models to generate loss-of-function signatures for selected targets. These genetic signatures serve as anchors for interpreting Chem-Seq profiles, enabling direct comparison between pharmacologic inhibition and genetic suppression of the same pathway. Where chemical and genetic perturbations converge transcriptionally, confidence in on-target activity is strengthened. Where they diverge, additional biology, compensatory signaling, or off-target effects can be explored. What the platform reveals in patient-derived organoids Using patient-derived organoids preserves key aspects of tumor heterogeneity and 3D architecture that are often lost in standard cell lines. Across the presented NSCLC PDX organoid models, Chem-Seq generated highly reproducible transcriptional signatures. Compounds targeting shared pathways clustered together, even when structurally distinct, while strongly cytotoxic agents formed separate transcriptional groups. Dose-response profiling added an additional layer of insight. Increasing compound concentration revealed clear transitions between pathway-selective signatures and broader cytotoxic stress responses. This distinction is critical for prioritizing compounds that modulate their intended targets without inducing nonspecific toxicity. Functional-Seq knockdowns recapitulated chemical inhibition for several pathways, demonstrating strong cross-method concordance at the gene and pathway levels. In contrast, targets that failed to produce robust genetic or pharmacologic signatures highlighted limitations related to knockdown efficiency or pathway redundancy. Why integrated Chem-Seq and Functional-Seq matter Together, Chem-Seq and Functional-Seq provide a scalable, mechanism-resolved framework for drug discovery in clinically relevant models. This integrated approach supports: Mechanism confirmation and target engagement assessment Rapid prioritization of compounds with clean, pathway-specific signatures Early identification of off-target or cytotoxic liabilities Improved interpretation of phenotypic screening data Because the platform is compatible with large compound libraries and patient-derived organoids, it bridges the gap between discovery-scale screening and translational relevance. Looking ahead Following validation, this workflow is designed to scale further. Large-scale screening across diverse PDX organoid models and standard-of-care comparators will expand the reference space of transcriptomic signatures. Combined with machine-learning and generative AI approaches, these datasets can be used to predict compound response, guide structure-activity relationships, and even design novel compounds optimized for desired transcriptional outcomes. By integrating high-throughput transcriptomics, genetic perturbation anchoring, and patient-derived 3D biology, Chem-Seq and Functional-Seq offer a powerful foundation for next-generation oncology drug discovery and precision medicine. Want to explore the full data? This blog highlights the overall strategy and key insights, but the complete poster includes detailed workflows, clustering analyses, concordance metrics, and pathway-level results. Download the AACR 2026 poster to explore the full Chem-Seq and Functional-Seq dataset and figures.
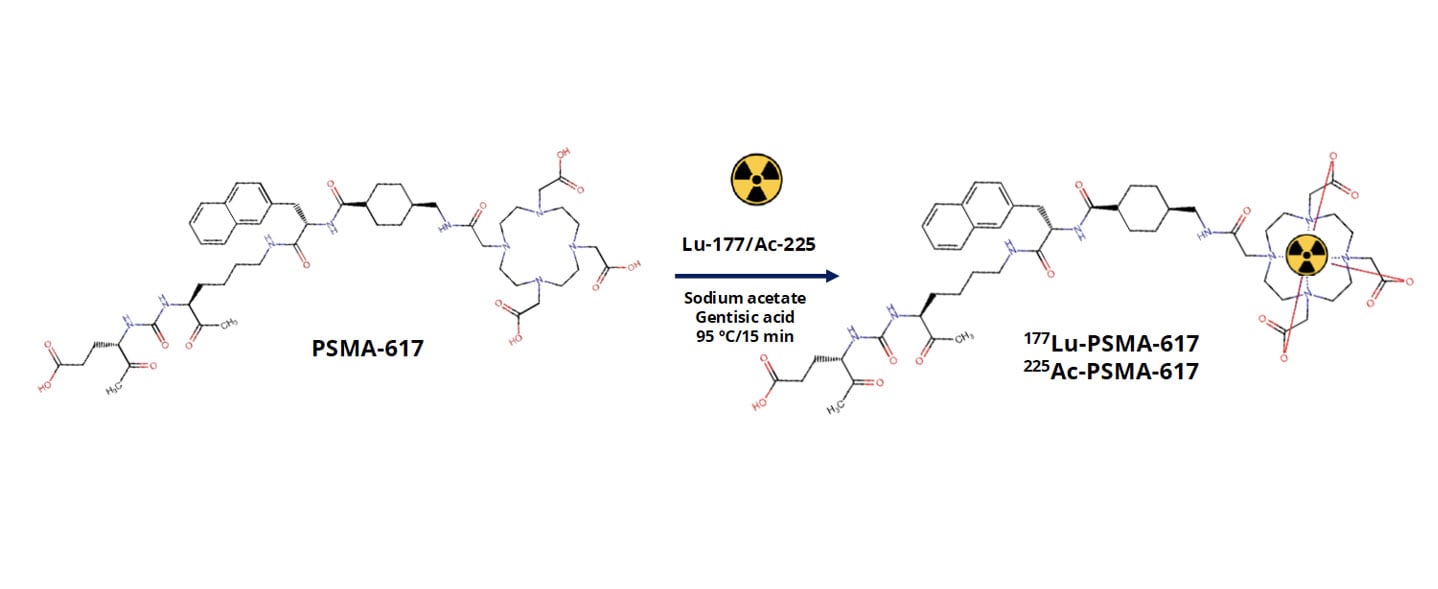
How Preclinical PDX Studies Help Guide PSMA Radioligand Development
Metastatic castration-resistant prostate cancer, or mCRPC, remains a major clinical challenge. While prostate-specific membrane antigen, PSMA, has emerged as a highly promising therapeutic target, differences in tumor biology, target expression, and radiopharmaceutical behavior continue to influence clinical outcomes. For PSMA-targeted radioligand therapies to reach their full potential, developers need preclinical models that reliably capture both tumor complexity and biodistribution behavior in vivo. At AACR 2026, we presented data showing how patientderived xenograft (PDX) models can be used to evaluate PSMA-targeted radiopharmaceuticals at multiple levels. This included radiochemistry quality, biodistribution, and tumor selectivity. The work focused on two clinically relevant agents, [177Lu]LuPSMA617 and [225Ac]AcPSMA617, tested across a panel of mCRPC PDX models with variable PSMA expression. Why PDX models are critical for radiopharmaceutical development Radiopharmaceutical therapies rely on far more than target binding alone. Successful translation depends on tumor uptake, retention, clearance from healthy tissues, and therapeutic index. Traditional cell line models fail to capture the architectural and microenvironmental features that shape these processes in patients. PDX models preserve the genetic heterogeneity and tissue organization of the original human tumor. This makes them particularly well suited for studying radioligand distribution and efficacy in vivo. In the context of PSMA-targeted therapies, PDX models allow us to directly evaluate how differences in PSMA expression and tumor biology influence radiotracer uptake in clinically relevant tumor models. Designing a clinically relevant PSMA radiopharmaceutical study In this study, we evaluated PSMA617 labeled with either lutetium177 or actinium225 across multiple prostate cancer PDX models representing metastatic, castration-resistant disease. The models spanned a range of PSMA expression levels, enabling us to assess how target expression directly relates to biodistribution. We first confirmed robust radiochemical performance. PSMA617 was efficiently labeled with both isotopes, achieving high radiochemical purity that met stringent quality standards. Establishing radiochemistry consistency is a critical first step before moving into in vivo evaluation, particularly for comparative studies involving different isotopes. How biodistribution studies inform target specificity and safety Following radiotracer administration, we conducted biodistribution analyses at defined time points to quantify uptake across tumors and major organs. Rather than relying on a single model, we assessed multiple PDXs to capture biologically driven variability. Across the panel, tumor uptake correlated strongly with PSMA expression levels. Models with high PSMA expression showed selective and robust radiotracer accumulation, while low-PSMA models demonstrated minimal uptake. This clear relationship confirms biological predictability and reinforces the value of molecularly annotated PDX panels when evaluating PSMA-targeted therapies. Importantly, although both [177Lu]LuPSMA617 and [225Ac]AcPSMA617 demonstrated tumor targeting, differences emerged in tumor-to-normal tissue ratios. These distinctions highlight why side-by-side preclinical evaluation is essential when selecting isotopes and dosing strategies for clinical development. Comparing beta- and alpha-emitting PSMA therapies One of the most important questions in PSMA radiopharmaceutical development is how different isotopes behave in vivo. Lutetium177 and actinium225 differ in emission properties, tissue penetration, and potential toxicity profiles. Our comparative biodistribution analyses showed that while both agents localize to PSMA-expressing tumors, [177Lu]LuPSMA617 demonstrated more favorable tumortotissue ratios across several organs. These findings provide early insight into how isotope choice may influence therapeutic window and tolerability. While alpha-emitters remain highly compelling for their potent cytotoxicity, data like this emphasize the importance of rigorous preclinical evaluation in clinically relevant models before advancing treatment strategies. Why this approach matters for radiopharmaceutical pipelines Radiopharmaceutical development is inherently multidisciplinary, integrating chemistry, biology, imaging, and therapeutic evaluation. PDX models provide a unifying platform where all of these components can be assessed in context. Using well-characterized mCRPC PDXs allows us to: Link PSMA expression directly to in vivo uptake Compare isotopes in clinically relevant tumor models Evaluate tumor selectivity alongside therapeutic response Reduce translational uncertainty before clinical studies This kind of integrated preclinical strategy is especially valuable as PSMA-targeted therapies continue to expand into earlier disease settings and combination regimens. Want to see the full dataset? This blog highlights key findings, but the full study includes detailed radiochemistry validation, biodistribution data, tumor-to-tissue ratios, and efficacy results across multiple PDX models. Download our AACR 2026 poster to explore the complete results and analyses.
How to Use PDX Models to Advance Folate Receptor-Alpha ADC Development in Ovarian Cancer
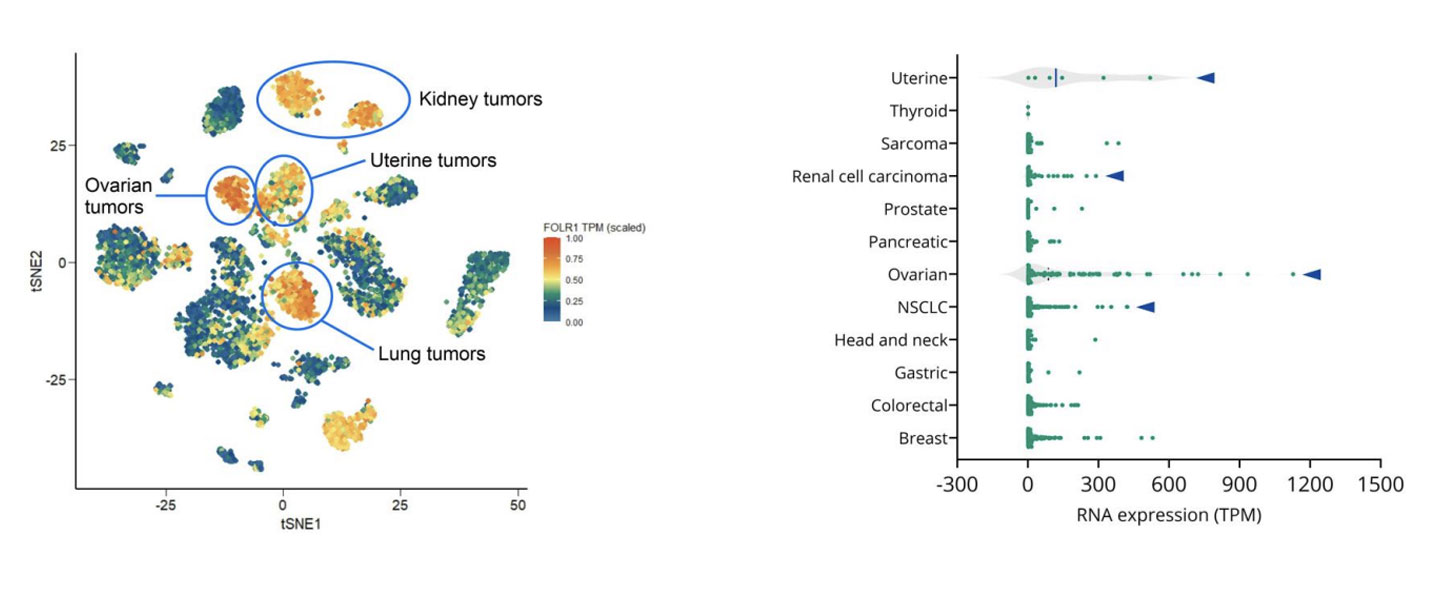
Epithelial ovarian cancer remains one of the deadliest gynecologic malignancies. Although many patients initially respond well to platinum-based chemotherapy, relapse is common, and outcomes for platinum-resistant disease have seen little improvement over the past decade. With limited treatment options and a shortage of robust predictive biomarkers, there is a clear need for more precise therapeutic strategies that can better match patients to targeted therapies. One promising area of progress is antibody-drug conjugates, or ADCs. These therapies are designed to deliver potent cytotoxic agents directly to tumor cells by targeting tumorassociated antigens. In ovarian cancer, folate receptoralpha, or FRα, has emerged as an especially compelling target due to its high expression in ovarian tumors and comparatively limited expression in normal tissues. At AACR 2026, we shared new data showing how patient-derived xenograft, or PDX, models can be used to evaluate FRα-targeted ADCs, explore variability in response, and begin to uncover potential mechanisms of resistance. This work highlights how clinically relevant PDX models, combined with molecular profiling, can support smarter ADC development decisions earlier in the pipeline. Why FRα is an attractive ADC target in ovarian cancer FRα is a transmembrane glycoprotein involved in folate transport and cellular metabolism. While healthy tissues show limited FRα expression, ovarian tumors, particularly high-grade serous carcinomas, often express it at high levels. This contrast makes FRα well suited for ADC approaches that aim to maximize tumor-specific activity while minimizing offtarget toxicity. Mirvetuximab soravtansine-gynx, marketed as Elahere®, is an FRα-targeted ADC recently approved for patients with FRα-positive, platinum-resistant ovarian cancer. However, clinical experience has shown that FRα expression alone does not reliably predict which patients will respond to treatment. Understanding this response heterogeneity is one of the key challenges in optimizing ADC development. How we select the right PDX models to reflect patient biology To address this challenge, we leveraged our gynecologic cancer PDX collection, which includes models derived from both treatment-naïve and heavily pretreated ovarian cancer patients. Using integrated RNA sequencing and immunohistochemistry, we assessed FRα expression across ovarian PDXs and compared these patterns with patient data. Consistent with clinical observations, ovarian cancer PDXs showed some of the highest FOLR1 expression levels across tumor types. Importantly, RNAbased expression data strongly correlated with membrane-specific FRα protein expression. This confirms that multiple complementary methods can be used to select biologically appropriate models for FRαtargeted ADC screening. Rather than relying on a single cutoff, this approach allows us to build PDX cohorts that better reflect the biological diversity seen in the clinic. What PDX efficacy studies reveal about ADC response Selected ovarian PDX models were treated with mirvetuximab soravtansine using a clinically relevant dosing schedule. As expected, models lacking FRα expression did not respond to treatment, reinforcing the importance of FRα as a prerequisite for activity. Among FRα-positive models, however, responses varied widely. Some tumors showed meaningful growth inhibition, while others demonstrated limited or no response despite high FRα expression. This mirrors what is seen in the clinic, where only a subset of FRαpositive patients achieves durable benefit from treatment. These results highlight a critical lesson for ADC development. Target expression is necessary, but it is rarely sufficient on its own. Using molecular analysis to explore resistance mechanisms To begin understanding why FRα-positive tumors respond differently, we compared responding and non-responding PDX models using transcriptomic and pathway-level analyses. Rather than focusing on single genes, we looked at broader biological processes that may influence sensitivity or resistance to ADC therapy. Clear differences emerged between sensitive and resistant tumors. In particular, pathways related to intracellular transport and drug efflux were enriched in non-responding models. Several of these pathways involve ATP-binding cassette transporters, which are well known for their role in multidrug resistance. While exploratory, these findings suggest potential mechanisms by which tumor cells may evade ADC-delivered payloads, even when the target antigen is present. This type of insight can help guide biomarker development and inform rational combination strategies. How we model acquired resistance using PDXs Primary resistance is only part of the challenge. In many patients, response to ADCs is followed by progression under treatment. To study this, we are actively generating PDX models from patients who initially responded to ADC therapy and later relapsed. In the AACR study, one such PDX was derived from a patient who had responded to mirvetuximab soravtansine before progressing. When re-challenged in vivo, this model showed resistance to treatment, suggesting that clinically acquired resistance mechanisms can be preserved in PDXs. Models like this provide powerful tools for studying resistance biology and evaluating nextgeneration therapies designed to overcome it. Why PDXs matter for ADC development Taken together, these findings show how PDX models can support ADC development beyond basic efficacy screening. When combined with molecular and bioinformatics analyses, they can help: Improve patient stratification beyond single biomarkers Reveal biological drivers of heterogeneous response Enable investigation of intrinsic and acquired resistance Support the development of next-generation ADCs and combination approaches As ADC pipelines continue to expand, integrating clinically representative PDX models early in development can improve translation and, ultimately, patient outcomes. Want to explore the full data? This blog highlights key insights, but the full dataset includes detailed efficacy results, pathway analyses, and figures that provide additional depth. Download our AACR 2026 poster to explore the complete results and analyses.
Understanding Sensitivity and Resistance to Pan-RAS(ON) Inhibition Across KRAS-Mutant Tumors
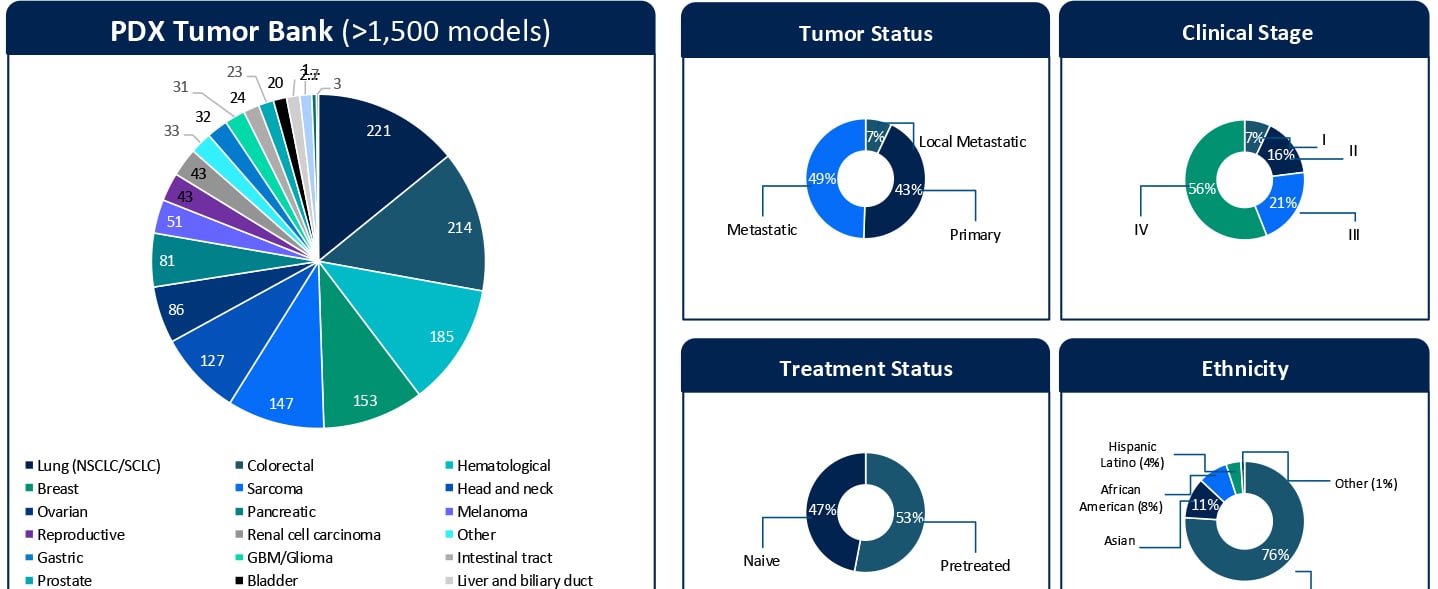
Understanding Sensitivity and Resistance to Pan-RAS(ON) Inhibition Across KRAS-Mutant Tumors KRAStargeted therapies are entering a new phase of clinical relevance. Daraxonrasib, a next-generation pan-RAS(ON) inhibitor designed to target a broad spectrum of KRAS G12X mutations, has shown impressive clinical activity. In a recent Phase III RASolute 302 trial for second-line metastatic pancreatic ductal adenocarcinoma (PDAC), daraxonrasib (RMC-6236) nearly doubled the median overall survival for patients to approximately 13.2 months, compared with 6.7 months for standard chemotherapy.1 These results highlight the rapid progress in this space. At the same time, clinical development is expanding KRAS-targeted therapies into earlier lines of therapy. Daraxonrasib is currently being evaluated in first-line settings, both as monotherapy and in combination approaches. While both clinical and preclinical data demonstrate activity across multiple tumor types, variability in response and the emergence of resistance remain key challenges that limit durable benefit. Additionally, the molecular determinants that define sensitivity, primary resistance, and acquired resistance remain incompletely understood. Addressing these gaps in our understanding of the molecular drivers of sensitivity and resistance is critical not only for patient selection but also for informing rational combination and sequencing strategies in an evolving treatment landscape. A Translational Platform to Study KRAS Biology in Context To better understand these response patterns, this study leveraged Champions Oncology’s low-passage, clinically annotated Patient-Derived Xenograft (PDX) Tumor Bank. With more than 1,500 models and extensive molecular and treatment history annotation, this platform captures both tumor heterogeneity and clinically relevant treatment context. Importantly, approximately half of the models represent pretreated disease, reflecting real-world patient populations. The translational relevance of this platform is supported by prior clinical correlation analyses performed using Champions’ low-passage PDX models. In a study by Izumchenko et al., treatment responses observed in these models were directly compared with clinical outcomes in matched patients across 92 individuals and 129 therapies.2 Strong concordance between the PDX and patient response was observed, with a positive predictive value of approximately 85% and a negative predictive value of approximately 91%. These findings establish Champions’ low-passage PDX models both as a tool for biological exploration and a clinically validated system for informing translational and drug development decisions. In this study, more than 50 KRAS-mutant PDX models were screened across non-small cell lung cancer, colorectal cancer, and pancreatic cancer (PDAC). The cohort included diverse KRAS alleles, co-occurring genomic alterations, and varying treatment histories, which allowed treatment response to be evaluated within a realistic molecular and clinical framework. Heterogeneous Responses Reflect Tumor-Specific Context Daraxonrasib treatment resulted in a broad spectrum of responses, ranging from strong disease regressions to progressive disease. Response variability was not solely explained by KRAS mutation status alone but was influenced by the broader genomic context of each tumor. For example, alterations such as MYC amplification were observed in both responding and non-responding models, suggesting that primary resistance is context-dependent and likely driven by multiple interacting genomic factors, rather than single genomic features. These findings highlight the importance of distinguishing between intrinsic resistance mechanisms and those that emerge under therapeutic pressure. Probing Primary Resistance in KRAS G12D Pancreatic Models Given the clinical relevance of KRAS G12D in pancreatic cancer, a focused analysis was performed in this subset. Differential gene expression analysis comparing responders and non-responders identified candidate markers associated with response, including MSLN and CYP24A1. While these findings are exploratory and based on a limited sample set, they provide an initial framework for stratifying tumors and generating hypotheses around mechanisms of intrinsic resistance. Ongoing work is expanding these analyses through deeper multi-omic integration, including whole exome sequencing (WES), RNA sequencing, proteomics, phosphoproteomics, and surface proteomics. These datasets will be interpreted in the context of Champions’ clinically annotated models, which include detailed treatment histories, to better understand the biological drivers of response and resistance and to support more robust biomarker development. Modeling Acquired Resistance Under Continuous Drug Pressure Selected PDX models were treated continuously beyond their initial response to investigate how resistance evolves over time. A particularly illustrative example is model CTG-2473, a KRAS G12D pancreatic cancer model derived from a tumor collected after first-line gemcitabine treatment. This model reflects a clinically relevant treatment sequence where patients, following standard chemotherapy, would be transitioned to KRAS-targeted therapies such as daraxonrasib. Therefore, treatment of CTG-2473 with daraxonrasib can model response and resistance in a post-chemotherapy setting that mirrors emerging clinical practice. Under continuous treatment pressure, this model demonstrated initial tumor regression followed by relapse, which is consistent with the development of acquired resistance observed in patients. This creates a powerful framework for translational studies. Experiments can be designed in the parental setting to evaluate therapeutic switching upon relapseto assess next-line agents or rational combination strategies. In parallel, Champions Oncology is actively developing a fully resistant subline through serial passaging under sustained drug pressure. This will enable deeper molecular characterization of acquired resistance mechanisms and provide a robust system for testing strategies for overcoming resistance. Together, these models establish a framework not only to observe resistance but also to actively model clinically relevant treatment sequencing scenarios. Why Clinically Annotated PDX Models Matter A defining strength of this work is the integration of molecular profiling, treatment response, and detailed clinical annotation within a single experimental system. Unlike conventional preclinical models, Champions’ PDX platform enables analysis of tumor behavior in the context of prior therapies, disease progression, and patient-specific treatment history. This is particularly important for targeted therapies such as KRAS inhibitors, where response and resistance are shaped by both molecular context and prior treatment. When combined with multi-omic datasets, including WES, RNA sequencing, proteomics, phosphoproteomics, and surface proteomics, these clinically annotated models provide a systems-level understanding of tumor biology. This supports the identification of candidate biomarkers and the development of mechanistically informed combination and sequencing strategies. Importantly, this integrated approach allows researchers to move beyond static response measurements tto capture how tumors dynamically adapt under therapeutic pressure and how these adaptations can be therapeutically targeted. From Mechanism to Strategy Taken together, these data demonstrate that response to pan-RAS(ON) inhibition is shaped by tumor-specific molecular context and dynamic adaptation under treatment. Understanding both primary and acquired resistance will be essential for translating KRAS-targeted therapies into durable clinical benefit. The full dataset, including detailed response profiles, genomic annotations, and resistance modeling, is presented in the accompanying poster. Download the Full Poster To explore the complete results, figures, and molecular analyses, including responder and non-responder comparisons and acquired resistance models, download the full poster here. References Daraxonrasib Demonstrates Unprecedented Overall Survival Benefit in Pivotal Phase 3 RASolute 302 Clinical Trial in Patients with Metastatic Pancreatic Cancer | Revolution Medicines [Internet]. Revolution Medicines. 2026. Available from: https://ir.revmed.com/news-releases/news-release-details/daraxonrasib-demonstrates-unprecedented-overall-survival-benefit Izumchenko E, Paz K, Ciznadija D, Sloma I, Katz A, Vasquez-Dunddel D, et al. Patient-derived xenografts effectively capture responses to oncology therapy in a heterogeneous cohort of patients with solid tumors. Annals of Oncology. 2017 Oct;28(10):2595–605.
Clinically Relevant 3D Tumor Models for HER2-Targeted ADC Development
Expanding the Preclinical Toolbox with a Novel Patient-Derived Organoid Platform The human epidermal growth factor receptor 2 (HER2) plays a central role in regulating cell growth, division, and survival. When overexpressed, it acts as an "on switch", driving accelerated cell growth and uncontrolled tumor spread. This biology underlies the aggressive behavior observed in HER2-positive cancers, which is associated with higher recurrence rates and poorer patient outcomes. While genomic changes in HER2 are most frequently discussed in the context of breast cancer, HER2 alterations are also found in non-small cell lung, ovarian, colorectal, and pancreatic cancers. HER2-Targeted ADCs in Cancer Therapy HER2-targeted therapies have significantly changed the treatment landscape and brought new hope and better outcomes for patients. Among them, HER2-targeted antibody-drug conjugates (ADCs) have emerged as a particularly promising approach, combining the specificity of antibodies with the potency of cytotoxic payloads. These agents enable targeted delivery directly to HER2-expressing tumor cells while limiting off-target toxicity. FDA approved drugs in this class include trastuzumab emtansine (T-DM1, Kadcyla®) and trastuzumab deruxtecan (T-DXd, Enhertu®), both of which have demonstrated meaningful clinical responses (Figure 1). A growing number of next-generation HER2 ADCs are now in development. Figure 1. FDA approved HER2-targeted ADCs trastuzumab emtansine (T-DM1, Kadcyla®) and trastuzumab deruxtecan (T-DXd, Enhertu®). Figure adapted from Joubert et al1. As ADCs become an increasingly important therapeutic modality, the limitations of conventional preclinical models have become magnified. Drug developers need better, more physiologically relevant and predictive preclinical models to support ADC development, ones capable of reproducing tumor architecture, heterogeneity, and treatment response. Limitations of Traditional 2D Preclinical Models for ADC Evaluation Despite their widespread use, traditional 2D cell culture models have well-recognized drawbacks with respect to evaluating targeted therapies. They often fail to capture key biological features of tumors and inadequately replicate the complex tumor microenvironment (TME) that influences drug response in patients. Target accessibility, receptor density, internalization kinetics, and payload penetration are all influenced by three-dimensional tumor structure, features which are largely absent in 2D cultures. As a result, 2D assays may overestimate drug potency or fail to distinguish on-target activity from nonspecific cytotoxicity, reducing their predictive value for clinical translation. In contrast, growing tumor cells as 3D organoids allows for a spatially organized structure that more closely resembles the in vivo microenvironment and architecture of patient tumors. Champions has developed a scalable 3D screening platform based on HER2-positive patient-derived xenograft organoids (PDXOs). Derived from well-characterized PDX models, our ex vivo 3D organoids maintain clinically translatable HER2 expression, providing a relevant and predictive platform for evaluating the efficacy of HER2-targeted ADCs. Validation of HER2-Positive PDXO Models HER2-positive ex vivo PDXO breast cancer models generated from our TumorGraft3D (CTG-3D) platform were evaluated for HER2 expression as well as functional responses to the clinically approved HER2-targeted ADCs, trastuzumab emtansine (T-DM1) and trastuzumab deruxtecan (T-Dxd). The workflow is shown in Figure 2. Figure 2. Workflow overview to evaluate HER2-targeted ADCs using breast cancer PDXO models. HER2 Expression Immunohistochemical (IHC) analysis of PDXO tissue blocks demonstrated complete and intense membrane staining (HER2 score 3+) across HER2-positive models. Consistent with these findings, flow cytometric analysis of enzyme-dissociated organoids identified HER2-positivity in 61.8% to 90.1% of cells. In contrast, the HER2-negative control PDXO model showed no detectable HER2 staining by IHC and negligible HER2 expression by flow cytometry, confirming assay specificity and the absence of nonspecific background signal. A subset of the results is shown in Figure 3. Figure 3. HER2 expression assessed by IHC and flow cytometry analysis. A) HER2-positive PDXO and B) HER2-negative PDXO model. Together, the IHC staining and flow cytometry results are consistent with the clinical annotation and indicate that HER2 expression is maintained relatively homogeneously in breast cancer PDXO models. Response to HER2-Targeted ADCs After confirmation of HER2 expression, the PDXO models were evaluated for their response to two clinically approved HER2-targeted ADCs, trastuzumab deruxtecan (T-DXd) and trastuzumab emtansine (T-DM1). Both ADCs were tested against HER2-positive and HER2-negative breast cancer PDXOs and reference breast cancer cell lines. In addition to intact ADCs, corresponding cytotoxic payloads (Exatecan and DM1), naked antibody (trastuzumab), and—for T-DXd—an isotype-payload conjugate (IgG-DXd) were evaluated to distinguish potency, selectivity, and target-dependent activity. Drug response was measured using the CellTiter-Glo® viability assay following six days of incubation with the ADCs and control treatments. Figure 4. Representative dose-response curves (IC₅₀). A) HER2-positive PDXO model comparing exatecan (payload) and trastuzumab deruxtecan (T-DXd) and B) control naked antibody trastuzumab and isotype-payload conjugate IgG-DXd. Across HER2-positive breast cancer PDXO models and control cell lines, ADCs demonstrated greater efficacy and specificity compared with naked antibody and isotype-payload controls (Figure 4, and data not shown). Full datasets across additional PDXO models are presented in the accompanying AACR 2025 poster. These results highlight the utility of PDXO models derived from Champions’ CTG-3D platform for evaluating next-generation ADCs and discriminating between payload-driven cytotoxicity, nonspecific conjugate effects, and HER2-targeted ADC activity. To further quantify differential treatment responses, normalized Area Under the Curve (∆AUC) values were calculated to capture response differences between isotype controls and ADCs across the full concentration range. HER2-positive breast cancer cell lines and PDXO models exhibited higher ∆AUC values (approximately 30–50%, data not shown), indicating stronger ADC activity relative to controls. In contrast, HER2-negative models showed substantially lower ∆AUC values (~20%), consistent with reduced ADC activity in the absence of target expression (Figure 5). Figure 5. Normalized ∆AUC. The response difference between isotypes and ADCs is calculated as: [AUC of Isotype] – [AUC of ADC] / [AUC of Isotype ] x 100% Collectively, these findings demonstrate that breast cancer PDXO models generated from Champions’ CTG-3D platform reproduce expected response patterns to FDA-approved HER2-targeted ADCs with known pharmacological profiles, supporting their utility as a predictive ex vivo platform for ADC development. Key Takeaways Champions’ CTG-3D platform is a biologically relevant ex vivo 3D platform that can support critical preclinical decisions, including payload comparison and mechanism-based screening, enabling ADC candidates to be screened and prioritized before more expensive in vivo evaluation and clinical development is undertaken. For drug developers, this translates into greater confidence in candidate selection, improved translational relevance of preclinical data, and reduced risk of late-stage attrition of potentially viable candidates. By enabling better-informed decisions earlier in the development process, the CTG-3D platform helps accelerate the advancement of safer and more effective ADC-based therapies. To explore the full datasets in more detail, download the full poster presented at AACR 2025 here. References Joubert, Nicolas, Alain Beck, Charles Dumontet, and Caroline Denevault-Sabourin. 2020. “Antibody-Drug Conjugates: The Last Decade.” Pharmaceuticals (Basel, Switzerland) 13 (9). https://doi.org/10.3390/ph13090245.
Evaluating a Novel GCN2 Inhibitor for Acute Myeloid Leukemia (AML) Treatment Using Patient-Derived Models
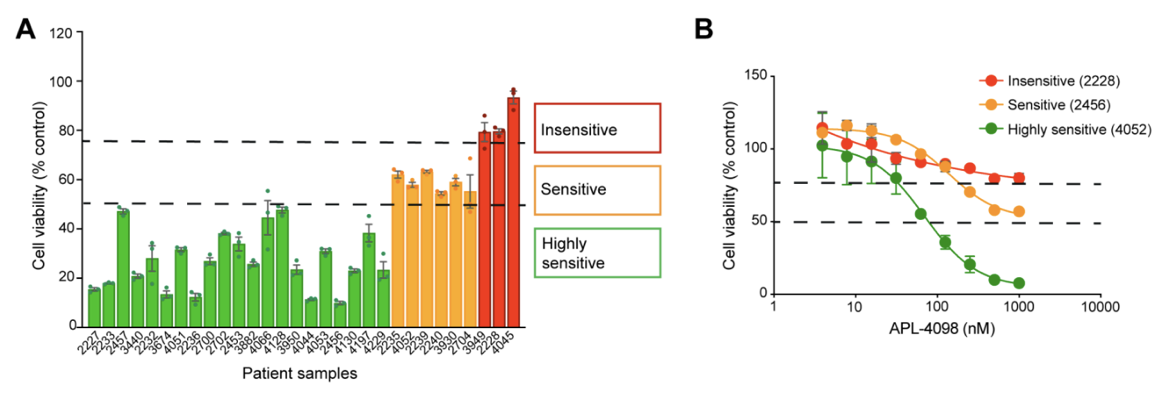
The AML Therapeutic Landscape Relapse remains one of the greatest challenges in treating Acute Myeloid Leukemia (AML). While targeted therapies have improved outcomes for some patients, high relapse rates and limited durable treatment options mean that many patients still face poor prognoses. One pathway gaining attention is the Integrated Stress Response (ISR), a highly conserved protective signaling network that helps cells adapt to nutrient deprivation, oxidative stress, and other metabolic stressors. Many tumor types, including AML, depend heavily on this stress-response system to survive and proliferate. Within this pathway, the kinase GCN2 (General Control Nonderepressible 2) serves as a key sensor of amino acid deprivation and metabolic stress, helping cancer cells survive under unfavorable conditions where lack of exogenous nutrients may disturb intracellular homeostatic balance. Disrupting this survival mechanism has emerged as a promising strategy to selectively target cancer cells and improve treatment outcomes. A recent study explored a novel and selective inhibitor of the stress-response kinase GCN2, APL-4098, as a potential therapeutic approach for AML1. Ex Vivo Evaluation of APL-4098 in Patient-Derived AML Cells Román-Trufero et al. first characterized the anti-leukemic activity of APL-4098 in ex vivo primary patient AML samples using Champions Oncology’s AML VitroScreen platform. A cohort of 30 patient-derived samples was treated with increasing concentrations of the compound and cell viability was measured using a luminescence-based viability readout to determine how effectively the drug inhibited leukemia cell growth. From these results, dose–response curves and IC50 values were generated to quantify drug potency across the cohort. The results revealed strong inhibitory activity across the patient cohort with 70% of samples (21/30) showing a ≥50% reduction in cell viability, indicating high sensitivity to the GCN2 inhibitor. An additional 20% of samples demonstrated some sensitivity, with a 25–50% reduction in viability. Finally, 10% of the samples showed less than 25% decrease in viability and were categorized as insensitive (Figure 1). Figure 1. APL-4098 cytotoxic activity ex vivo. (A) Effect of 500 nM APL-4098 on the viability of patient-derived AML samples after 72h treatment. Values for each specimen are expressed relative to each sample’s matched vehicle-treated control (normalized to 100%) and presented as mean ± SEM, n=3. (B) Selected APL-4098 dose-response curves of highly sensitive (green), sensitive (orange), and insensitive (red) patient-derived samples. Notably, responses were observed across diverse genomic backgrounds, with no clear correlation between common AML mutations and sensitivity. Cells from pre-treated or relapsed patients also responded, suggesting that GCN2 inhibition may be effective even in difficult-to-treat AML. This ex vivo screening step provided critical insight into which patient samples were most sensitive, laying the groundwork for subsequent in vivo evaluation in patient-derived xenograft (PDX) models. In Vivo Evaluation of APL-4098 in AML Patient-Derived Xenografts Following ex vivo screening, the anti-leukemic activity of APL-4098 was further assessed in Champions’ patient-derived xenograft (PDX) AML models. Human AML cells from a patient sample that showed ex vivo sensitivity to APL-4098 were implanted into immunodeficient NOG mice via tail vein injection, allowing leukemia to engraft in the bone marrow. Engraftment of human AML cells in the bone marrow was monitored in surrogate animals using flow cytometry, and once engraftment reached ~20% human CD45+ cells, mice were randomized into four treatment groups: Vehicle control (DMSO) APL-4098 alone (15 mg/kg once daily) Venetoclax alone (100 mg/kg once daily), as a standard-of-care comparator APL-4098 + venetoclax combination Treatments were administered daily for 19 days, and leukemia burden was evaluated by analyzing multiple AML subpopulations, including blasts, progenitors, and leukemia stem cells (LSCs), using multicolor flow cytometry (Figure 2). This allowed researchers to evaluate both single-agent activity and potential synergy with venetoclax in a clinically relevant AML model. Figure 2. Effect of APL-4098, Venetoclax and the combination of both in vivo in an AML PDX model. Bone marrow from AML-engrafted animals was assessed for (A) the percentage of viable human CD45+. (B) the number of viable blasts (CD34+/CD33+/-). (C) the number of viable AML progenitors (CD34+/CD38+) (D) the number of viable LCSs (CD34+/CD38-). In all graphs, data shown as mean ± SEM, statistical significance determined by one-way ANOVA. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. DMSO, n=6; APL-4098, n=7; Venetoclax, n=8, APL-4098 + Venetoclax, n=7. The results showed APL-4098 as a monotherapy had little to no effect on bulk leukemic burden, but preferentially targeted and reduced the LSC-enriched compartment, a niche generally considered primarily responsible for relapse initiation after bulk eradication of leukemic blasts2 Importantly, the combination of APL-4098 and Venetoclax, the standard of care BCL-2 inhibitor in AML patients, produced a pronounced synergistic response, achieving a 97% to 99% reduction across all leukemia subpopulations analysed. Mechanistic studies, including RNA sequencing and metabolic analyses, suggested that APL-4098 exerts its anti-leukemic activity by suppressing mitochondrial respiration, triggering the mitochondrial unfolded protein response, and inducing metabolic stress in AML cells. Together, the researchers demonstrated APL-4098 effectively induces cytotoxicity both ex vivo and in vivo, with preferential effect against the LSC subpopulation, a key driver of patient relapse. While APL-4098 shows potential as a monotherapy agent in the preclinical setting, the potent synergistic interaction between APL-4098 and Venetoclax highlights a potential combination therapy for AML that merits further non-clinical and clinical exploration. This study demonstrates the utility of Champions Oncology’s patient-derived platforms for preclinical and translational AML research, providing a framework to evaluate drug efficacy and explore new therapeutic strategies. To learn more about this study, download the full publication here Explore the Champions Oncology website to discover more about our hematological cancer models and testing capabilities References Román-Trufero M, Whitlock G, Seydoux C, et al. A novel potent and selective GCN2 inhibitor, APL-4098, has anti-leukemic activity through dysregulation of mitochondrial function. Clin Cancer Res. 2026. Hansen Q, Bachas C, Smit L, Cloos J. Characteristics of leukemic stem cells in acute leukemia and potential targeted therapies for their specific eradication. Cancer Drug Resist. 2022.