
Trends in Oncology
Advancing the Battle Against Mantle Cell Lymphoma
Mantle cell lymphoma (MCL) is a rare and aggressive non-Hodgkin’s lymphoma (NHL) that originates in B cells and occurs in secondary follicles of the lymph nodes.1 MCL accounts for roughly 6% of all lymphomas diagnosed annually, and despite its relatively low incidence, the aggressive course of MCL means it can quickly become a life-threatening disease. Understanding the origin and progression of MCL is crucial to identify new targets for drug development studies. Understanding Mantle Cell Lymphoma Pathology Over the last decade, our understanding of MCL pathogenesis has evolved from a single definition to a consensus that MCL development and progression is related to a range of molecular events. Early on MCL was defined as a pathognomonic chromosomal translocation t(11;14)(q13;q32) causing a mutation in the CCND1 gene, resulting in the over-expression of Cyclin D1.1, 2 Under normal conditions, Cyclin D1 is heavily regulated and modulates cell cycle transition from G1 phase to S phase.1 However, overexpression of Cyclin D1 activates cyclin-dependent kinases (CDK) 4 and 6 which later deactivate the retinoblastoma protein (Rb), a cell cycle inhibitor. This series of events accelerates cell cycle progression from G1 to S phase in many cell types, including B-cells.3 The progression of this process induces uncontrolled B-cell proliferation causing an enlargement of lymph nodes and immune system dysfunction that eventually spreads to critical organs. Fewer Mutations Raise More Questions Interestingly, Cyclin D1 mutations are not found in all MCL cells, suggesting other molecular actors, such as transcription factors, are involved. In 90% of MCL patients, transcription factor SOX11 is over-expressed in MCL cells with and without mutated CCND1. While SOX11’s role in MCL is not well understood, studies suggest that PAX-5, a transcription factor that regulates B-cell development and differentiation, is activated by SOX11.4 The overexpression of SOX11 that is commonly seen in MCL patients can therefore lead to reduced B-cell differentiation and increased B-cell antigen signaling.3, 5 Identifying Treatment Targets in Mantle Cell Lymphoma B-cell proliferation and differentiation rely on B-cell receptor (BCR) signaling and are activated in response to antigen binding.6 Using the framework for CLL pathogenesis, preliminary MCL-BCR research has found that BCR signaling is highly active in MCL patients and intentional BCR activation resulted in an increase of BCR signaling in MCL cells.7 Additionally, murine studies have found SOX11-overexpressing B-cells have high levels of BTK, a key enzyme involved with BCR signaling, resulting in proliferation which suggests SOX11’s deeper role in MCL.7 Initial MCL treatment includes R-CHOP therapy, however, many MCL patients are refractory or become resistant creating a need for additional therapies. As with CLL, BCR signaling plays a critical role in MCL progression shifting treatment protocols towards BTK inhibitors (BTKi) like ibrutinib, approved in 2013.1, 3 Ibrutinib is metabolized in the liver via CYP3A and CYPRD6 and irreversibly binds to cysteine residue 481 found on the active site of BTK. While this targeting strategy effectively blocks BTK signaling limiting MCL cell development, ibrutinib has significant off-target consequences, namely IL-2 inducible kinase (ITK), which sparked the development of zanubrutinib.8 Zanubrutinib was approved for use in MCL patients and though it has a similar mechanism of action to ibrutinib, its reduced affinity for ITK, a major T-cell and natural killer (NK) cell regulator,9 makes it a highly selective and potent BTKi with improved overall response rate relative to ibrutinib.8 Resistant Mantle Cell Lymphoma and Future Therapeutic Strategies Unfortunately, 32% of MCL patients become resistant to BTKi due to BTK mutations.10, 11 As a result, combination therapies such as ibrutinib combined with cirmtuzumab (a novel anti-ROR1 monoclonal antibody) or with obinutuzumab and venetoclax have been used to treat resistant MCL patients. 11 These therapeutic combinations have shown excellent remission results paving the way for other combination therapies, such as acalabrutinib, rituximab, and bendamustine. 11, 12 Notably, a phase 1 study investigating acalabrutinib, rituximab, and bendamustine achieved an 85% overall response rate which has led to a larger, ongoing Phase 2 study (NCT04115631). Additionally, novel MCL-1 inhibitors are under development in preclinical studies.13 Recently, the FDA has approved the first CAR T cell therapy for relapsed/refractory MCL, brexucabtagene autoleucel (Tecartus).14 Tecartus has shown durable response at the 3-year follow-up even in high-risk patients (NCT02601313), with sustained survival and a 67% complete response rate.15 Understanding the pathophysiology of naïve and BTKi resistant MCL is crucial to developing curative MCL therapies. While standard of care treatments like ibrutinib and zanubrutinib have had some success, additional studies investigating strategic combination therapies are underway providing hope for patients battling this disease. Champions supports your in vivo preclinical studies with low passaged MCL PDX models available for subcutaneous modeling and fully characterized with NGS data in 4 models CTG-3771, CTG-3772, CTG-3776 (shown to be rituximab and ibrutinib resistant) and CTG-3808. Preclinical hematological scientists need to evaluate their pipeline of therapeutic candidates in robust hematological screening platforms; Champions' VitroScreen platform can advance potential next-generation therapies into the clinic, generating additional options for MCL patients.
Exploring DRUG-seq: Revolutionizing RNA-seq in Oncology Research
Oncology research is a field that continuously demands cutting-edge technologies to unravel the complexities of cancer. Among these innovations, RNA sequencing (RNA-seq) has emerged as a vital tool, enabling us to study the transcriptome with unprecedented depth. However, a new star is on the rise in the realm of RNA-seq — DRUG-seq. This application dazzles oncology researchers and genomics professionals with its cost-effective approach, reduced bias, and high sample efficiency, bringing with it a promise to redefine how we investigate cancer at a molecular level.[1] Champ-seq is Champions's DRUG-seq platform. In this comprehensive exploration, we'll dissect the advantages of DRUG-seq, and map out its pivotal role in oncology research, shedding light on its unique applications and use cases in the fight against cancer. Unveiling DRUG-seq: The New Era in RNA-Seq RNA-seq, widely employed for quantifying gene expression, has been a game-changer in understanding the genetic mechanisms driving cancer. However, DRUG-seq takes this a step further, providing a powerful lens through which to examine the transcriptome under a spectrum of conditions, particularly those relevant to drug treatment responses. While traditional RNA-seq methods offer insights into the steady state of gene expression, DRUG-seq provides a dynamic view that captures the immediate and prolonged gene expression changes following drug administration. This level of detail is especially critical in oncology, where the intricate interplay of genetics, environmental factors, and treatment response paints a highly complex picture of disease progression and therapy outcomes. By enabling high-throughput profiling of the transcriptional response to drug compounds, DRUG-seq stands out as a catalyst for precision medicine, biomarker discovery, and the personalization of cancer treatment strategies.[1] The Advantages of DRUG-seq in Comparative Analysis Cost-Effectiveness One of DRUG-seq's most touted benefits is its cost-effectiveness. The methodology uses a smaller number of sequences to cover the transcriptome, thanks to its selective enrichment of pre-existing reference indices. This focus on specific gene regions, associated with drug response or otherwise, lowers the overall sequencing cost per sample, making large-scale comparative studies feasible within more restrained budgets. Sample Efficiency With oncology often facing constraints of sample availability, the high efficiency of DRUG-seq is a game-changer. It requires smaller amounts of starting material, which not only conserves precious samples but also aligns with the trend toward microsampling in emerging clinical research practices. DRUG-seq in Action: Enhancing Oncology Research and Development Drug Response Profiling An immediate application of DRUG-seq is in profiling the response of cancer cells to different compounds. By comparing the expression profiles pre- and post-treatment, researchers gain a comprehensive view of how drugs affect gene networks. This deep understanding underpins the development of more effective therapies by identifying compounds that selectively target critical pathways in specific cancer types.[2] Identifying Novel Drug Targets DRUG-seq empowers the hunt for new targets by revealing unsuspected links between gene expression patterns and drug effects. This insight into the cellular response can lead to the discovery of novel molecular targets that modulate sensitivity or resistance to treatment, providing fertile ground for the next generation of anti-cancer compounds. Biomarker Discovery Precision oncology heavily relies on the discovery and validation of biomarkers to predict patient outcomes and guide therapeutic decisions. DRUG-seq, with its ability to uncover gene signatures indicative of treatment response, plays a pivotal role in biomarker discovery, potentially leading to tests that can stratify patients for tailored treatment interventions.[1] Tumor Heterogeneity Cancer is not a single disease but a collection of disorders, each with its own molecular profile and behavior. DRUG-seq's power to unravel drug response within this context is invaluable, as it allows researchers to study how different cell populations within a tumor respond to treatment. This understanding of tumor heterogeneity can inform the development of combination therapies that target multiple facets of the disease. Conclusion: The Bright Future of high-throughput transcriptional profiling with Champ-seq in Oncology Research From cost-effectiveness and reduced bias to sample efficiency and rich data, Champ-seq provides a valuable addition to the oncologist's toolkit. Its applications span across drug and biomarker discovery, as well as understanding tumor heterogeneity, which is pivotal in the era of personalized medicine. As we have navigated the many advantages, it's clear that Champ-seq isn't just a new trend; it's a technological leap that can redefine the standard for RNA-seq applications in oncology research. By harnessing this tool, researchers can explore cancer treatments with unprecedented precision and efficacy, ultimately leading to improved patient outcomes and a more comprehensive arsenal against this formidable foe.

Developing Flow Cytometry Assays in Non-Human Primates (NHPs)
Non-human primates (NHPs) continue to be a valuable resource for preclinical research because of the similarities they share with humans. NHPs, especially rhesus macaques, are used in preclinical studies for evaluating new drugs or vaccines for safety and efficacy. Flow cytometry assays can be easily adapted to study cells from NHP. Consider these three factors if you are planning to adapt a flow cytometry assay for use with NHP samples. Staining panel prep: Like human subjects, PBMCs can be collected from Non-human primates and evaluated by flow cytometry. Many immune cell subsets, especially B and T cells, share similar phenotypes with their human counterparts. Human antibodies are most often used to stain NHP cells because of similarities shared between surface markers. Consider which cells of interest you may want to study and how existing antibodies used in flow cytometry may be used in your panel. Real-world variability: Many scientists are accustomed to working with an inbred mouse strain, which reduces background variability observed in experimental settings. In contrast, NHPs are outbred animals, and like humans, may display widely varying responses. As you develop your NHP protocols, consider how you will handle highly variable data, including flow cytometry data. These considerations will inform how many animals may be used in each group and how many cells are stained and evaluated in each sample. Metabolism and toxicity: NHPs share many physiological similarities to humans and are a valuable model for pharmacokinetic and pharmacodynamic measurements of experimental drugs and biologics. Toxicity testing can also be done in NHPs and together these data are critical to evaluating candidates in preclinical pipelines. Developing NHP protocols from inception to sample analysis requires working with experts who have experience writing protocols that are compliant with appropriate oversight committees, such as institutional animal care and use committee offices. Working with experts, such as contract research organizations, also assures that any NHP flow cytometry studies will satisfy any necessary regulatory compliance determinations related to drug and biological development.
Triplet Therapy for IDH1-Mutant AML Tumors
Acute myeloid leukemia (AML) is the most common leukemia among adults and has been challenging to treat with modern therapies.1, 2 This disease is highly heterogeneous and characterized by the rapid proliferation of undifferentiated myeloid cells that accumulate within the bone marrow. Several mutations and epigenetic abnormalities characteristic of AML have been targeted through chemotherapies or molecular inhibitors. Fortunately, the relentless pursuit of innovative, effective AML therapies has led to a deeper understanding of AML pathogenesis, such as the role of isocitrate dehydrogenase (IDH) mutations.3 IDH mutations have unique properties that, if properly targeted, may reshape AML patient outcomes and survival rates. Unstable Gene Expression The IDH family consists of three isoenzymes (IDH1, IDH2, and IDH3) and has an important role in the biosynthesis of metabolites involved in the tricarboxylic acid (TCA) cycle. Notably, IDH1 functions as a catalyst for reversible conversion of isocitrate to α-ketoglutarate in the cytosol as well as peroxisomes, yielding 1 NADPH. Downstream, α-ketoglutarate is reduced to D-2-hydroxyglutarate (D-2-HG) as part of the TCA cycle.3 Mutations in IDH1 are present in ~20% of AML patients and result from a single amino acid substitution at Arg132.4 This mutation induces an end-product shift that reduces α-ketoglutarate to R-2-HG instead. Unlike D-2-HG, R-2-HG competitively inhibits α-ketoglutarate-dependent enzymes, such as the TET family, and upon accumulation leads to impaired cellular differentiation and deregulation of DNA methylation. 3, 5 This alteration of DNA methylation and ultimately gene expression activates oncogenes and deactivates tumor-suppressor genes.6 The destabilizing features of IDH1 mutants have thus made this isoenzyme an interesting target for AML therapies. IDH Focused Therapies Since the identification of IDH mutations in 2008, the Food and Drug Administration (FDA) has approved two therapeutics for AML patients with IDH mutations, ivosidenib and enasidenib. Ivosidenib is a reversible, selective inhibitor that binds to the active site of mutated IDH1 to prevent the production of R-2-HG.7, 8, 9 Reduction in R-2-HG reportedly increases D-2-HG concentrations by 100x and restores DNA methylation and cellular differentiation in AML patients.10, 11 Though successful, resistance to ivosidenib and IDH inhibition has emerged, underlining the need for combination therapies that prevent IDH resistance.12 Triplet Therapy There are no triplet regimens currently approved for use in AML, however, clinical trial results have demonstrated successful remission in relapsed and naïve AML patients.13, 14 For instance, an ongoing phase Ib/II study examining ivosidenib and venetoclax with or without azacytidine has shown successful remission in patients with AML IDH1 mutations.13 The durable response to this therapy is especially promising, as the safety profile of this triplet therapy is similar to doublet therapies such as azacytidine + venetoclax and azacytidine + ivosidenib.13, 15 The median event-free-survival (EFS) for patients treated with this new triplet therapy was 36 months13, while previous azacytidine + ivosidenib combination treatments have achieved median EFS or 24 months.16 The Therapeutic Pipeline Prolonged EFS in AML patients receiving ivosidenib in combination with AML standards of care, such as venetoclax and azacytidine, has recently gained the attention of the European Commission (EC). In 2023, the EC announced the approval of ivosidenib in combination with azacitidine for AML patients with an IDH1 R132 mutation.18 Further, clinical studies evaluating the side effects and appropriate dosages of ivosidenib and venetoclax with or without azacytidine are underway. As more clinical data is released, access to more robust therapeutic treatments will become available to improve AML patient outcomes.
Providing Optimal Clinical Care for Cholangiocarcinoma Subtypes
Cholangiocarcinoma is a type of cancer that originates from cholangiocytes, the cells that constitute the bile ducts in the liver, which carry bile from the liver to the small intestine. It is a rare and aggressive cancer that can be categorized into different subtypes based on histological and molecular characteristics. Histologically, cholangiocarcinoma can be classified as intrahepatic, perihilar, or distal. Intrahepatic cholangiocarcinoma originates within the liver, while perihilar and distal cholangiocarcinoma develop in the ducts outside the liver. These subtypes have distinct clinical presentations and treatment approaches [1,2]. In this blog, discover the diverse nature of cholangiocarcinoma subtypes and learn about the specific clinical care required for each subtype. Histological Characteristics and Clinical Care Histological characteristics play a significant role in guiding clinical care for cholangiocarcinoma subtypes. Intrahepatic cholangiocarcinoma often presents as a solitary mass within the liver, while perihilar cholangiocarcinoma typically involves the bifurcation of the bile ducts. Distal cholangiocarcinoma commonly manifests as a tumor in the lower part of the bile duct near the small intestine. The histological features of each subtype influence the choice of diagnostic tests, surgical interventions, and other treatment modalities. For example, surgical resection is often the primary treatment option for intrahepatic cholangiocarcinoma, while perihilar cholangiocarcinoma may require a combination of surgery and liver transplantation. Distal cholangiocarcinoma may be treated with surgery, radiation therapy, or systemic chemotherapy. By understanding the histological characteristics of cholangiocarcinoma subtypes, healthcare professionals can provide appropriate clinical care to improve patient outcomes. Cholangiocarcinoma Subtype-Specific Therapies Each cholangiocarcinoma subtype requires specific therapies tailored to its unique characteristics. For intrahepatic cholangiocarcinoma, surgical resection is often the primary treatment approach, followed by adjuvant therapy such as chemotherapy or radiation therapy. Liver transplantation may be considered in selected cases. Perihilar cholangiocarcinoma, on the other hand, often requires more complex surgical interventions, such as liver resection combined with bile duct resection and reconstruction. Liver transplantation may be an option for patients with advanced disease or underlying liver disease. Distal cholangiocarcinoma may be treated with surgical resection, radiation therapy, or systemic chemotherapy. The choice of treatment depends on the extent of the tumor and the patient's overall health. By tailoring therapies to the specific cholangiocarcinoma subtype, healthcare professionals can optimize treatment outcomes and improve patient survival rates. Molecular Profiling and Treatment Strategies In addition to histological characteristics, molecular profiling has emerged as an essential tool for understanding cholangiocarcinoma subtypes and guiding treatment strategies. Molecular profiling involves analyzing the genetic and molecular alterations in cancer cells to identify potential targets for therapy. Advancements in molecular profiling techniques have allowed researchers to identify specific biomarkers and genetic mutations associated with cholangiocarcinoma subtypes. This information helps in developing targeted therapies that can effectively inhibit the growth and spread of cancer cells. Two targeted therapies are currently available for patients presenting tumors with FGFR2 fusions/rearrangements (infigratinib) or IDH1 (ivosidenib) mutation [3,4]. In general, treatment strategies for cholangiocarcinoma subtypes may include targeted therapies, immunotherapy, chemotherapy, and radiation therapy. Molecular profiling enables healthcare professionals to select the most appropriate treatment options based on the molecular characteristics of the tumor and the patient's overall health [5]. Future Directions in Cholangiocarcinoma Care The field of cholangiocarcinoma care is rapidly evolving, with ongoing research and advancements in treatment options. Efforts are being made to improve early detection methods for cholangiocarcinoma. Early diagnosis allows for timely intervention and increases the chances of successful treatment. Additionally, advancements in precision medicine and molecular profiling techniques hold promise for tailoring treatment plans based on the unique molecular characteristics of each patient's tumor. In conclusion, the diverse nature of cholangiocarcinoma subtypes necessitates specific clinical care for optimal treatment outcomes. Through a comprehensive understanding of histological and molecular characteristics, healthcare professionals can provide personalized therapies and contribute to ongoing research efforts aimed at improving cholangiocarcinoma care.
A Needle in a Haystack: Finding Rare AML Populations by Flow Cytometry
Hematologic malignancies include a wide array of lymphomas and leukemias that affect different immune cell subsets. Acute myelogenous leukemia (AML) is one of the most commonly occurring leukemias in adults and children. AML is a highly heterogeneous disease that can be caused by spontaneous gene mutations or chromosomal translocations, which result in the proliferation of dysfunctional myeloid cells. Cytogenetic and morphologic analyses have been the gold standard methods used in AML diagnosis. Still, flow cytometry-based protocols are becoming more widely used and validated as complementary diagnostic methods that can be coupled with these analyses to better guide treatment plans. Flow cytometry has also become an essential tool to understand AML progression and develop and evaluate novel therapeutics. Consider these aspects of flow cytometry-based analysis of AML for exploratory or preclinical research: Phenotype matters: Immunologists know a lot about what a normally developing myeloid cell should look like in terms of its immunophenotype. Changes in expression of different lineage markers correlate strongly with AML progression and are characterized by the presence of blasts (leukemic cells) in the bone marrow. Flow cytometry-based immunophenotyping offers researchers a rapid and sensitive method for detecting blasts at the onset of disease as well as monitoring changes in this population throughout an experimental therapy. Rare cells from precious samples: A major consideration of following AML progression is the ability to detect relatively rare cells in bone marrow aspirates. This type of sample may be relatively small and is typically used fresh for morphologic evaluation, but even a small volume of remaining aspirate can be used for flow cytometry-based methods that are sensitive and robust enough to detect blasts. Paired samples: Bone marrow cells can be analyzed along with peripheral blood using advanced flow cytometry methods that monitor the persistence of blasts and other leukemic subsets over time in these compartments. This type of analysis offers critical insight into the development of new therapeutics whether they are being evaluated in humanized mouse models or clinical trial participants. Flow cytometry continues to advance immuno-oncology research, especially for diseases involving the detection of rare cell populations. Consider working with preclinical and clinical flow cytometry experts to develop protocols for future immuno-oncology studies.
Enhancing CAR T Cell Therapy: Optimizing Preparation for Superior Results
Chimeric antigen receptor-mediated T cell (CAR T cell) therapies have revolutionized the treatment of hematologic malignancies and solid tumors. This therapy uses T cells, typically harvested from patients, that are engineered to express chimeric antigen receptors (CAR) specific to tumor cell antigens. CD19-targeting CAR T cell therapy was the first immunotherapy shown to effectively treat acute lymphoblastic leukemia[1], but a subset of patients relapse due to loss or poor engraftment of CAR T cells. Here we highlight advances in CAR T cell therapy to improve the quality of the immunotherapy product ex vivo for more effective responses in vivo. Culture Conditions T cells from peripheral blood mononuclear cells (PBMCs) are the primary cellular product used for CAR T cell therapy, but several steps must be carried out ex vivo to ensure that enough cells are made for therapeutic efficacy. The best treatment outcomes have been linked to high levels of CAR T cell engraftment and persistence upon transfer into a patient[2]. Ex vivo culture methods have been optimized to expand T cells, and cultures using less differentiated T cells, like stem cell memory T cells or naïve-like T cells, have been linked to better persistence upon transfer[3]. Recent studies are characterizing phenotypic features of T cells that preserve “stemness” upon ex vivo culture but still allow for expansion and expression of CAR T receptors. Reduced culture time has been one of the most effective methods for improving engraftment and antitumor responses but is limited by the number of cells yielded by this minimal manipulation process[4]. Biodistribution The successful engraftment and persistence of CAR T cells depend greatly on where cells migrate upon transfer into patients. Preserving stem cell-like features correlates with better engraftment, especially if donor-derived cells are unavailable and autologous cell sources must be used[5]. In vivo imaging studies have been very helpful in understanding CAR T cell dynamics and anti-tumor responses. Studies in mouse models have indicated that CAR T cells can get trapped in tissues, including the lungs, which can limit access to tumor targets[6]. Analysis of CAR T cells with tumor cells also revealed extensive functional heterogeneity, including CAR-T cells that can interact with tumor cells but not exert cytotoxic effector functions. A recent first-in-human study examined the biodistribution of radioisotope-labeled CAR T cells and confirmed that these cells rapidly distribute to tumor tissue but are taken up by the liver and spleen and can persist systemically for up to two weeks[7]. As new in vivo imaging studies are carried out, these insights will inform how CAR T cell products are made and delivered and are likely to improve treatment outcomes. Advances in ex vivo methods for CAR T cell preparation are already improving outcomes, and these methods are broadly applicable to donor-derived or autologous T cell products. In vivo imaging methods are also delineating characteristics of CAR T cells that improve tumor targeting or result in misdirected tissue homing. Future in vivo and ex vivo studies are well-poised to further advance CAR T cell applications.
Unlocking the Potential of CAR-T Therapy for Treatment of Multiple Myeloma
Multiple myeloma (MM) is the second most common hematological cancer[1] and results from a series of mutations that lead to malignant plasma cells in the bone marrow.[2] The survival of MM ranges from 5 to 7 years on average, although patients with high-risk MM, defined as inactive TP53 or 1q21 activation, have reduced survival of 2 years.[3, 4] Due to the complexity of this disease, MM remains incurable, however, the emergence of CAR-T (chimeric antigen receptor T-cell) therapy has provided hope for MM patients. Pathogenesis Under normal conditions, plasma cells secrete polyclonal protein or immunoglobulins comprised of one heavy chain and one light chain.[5] MM is derived from a series of mutations in plasma cells and clonal plasmacytes that instead produce monoclonal proteins (M-proteins) within the bone marrow (BM).[6] This disease typically progresses from monoclonal gammopathy of undetermined significance (MGUS) that later develops into smoldering multiple myeloma (SMM) – both of which are asymptomatic plasma cell disorders with no end-organ damage.[7] MGUS is defined as a lower percentage of M-protein (<3g/dl) while SMM is defined as M-protein concentration exceeding 3g/dl.[8] It is thought that initial mutation events seen in MGUS, and ultimately in MM, that lead to M-protein secretion occur during somatic hypermutation or class-switch recombination events, namely translocations involving IgH locus and IgL locus.[7] The results of these mutations form an abundance of dysfunctional plasma cells within the bone marrow leading to reduced immune system responses and organ damage. Therapeutics Standard of care treatment for newly diagnosed MM includes a cocktail of high-dose chemotherapy agents: bortezomib, lenalidomide, and dexamethasone.[9] These chemotherapeutic agents are effective at damaging tumor cell DNA as well as tumor microenvironment. Following this aggressive combination therapy, patients undergo allogeneic stem cell transplantation (ASCT) to restore blood cell production.[10, 11] Despite the success of these treatments, many MM patients experience graft-versus-host disease (GVHD) and/or relapse,[12] making it essential for new and innovative treatments to emerge. One treatment that has shown promise is chimeric antigen receptor T cell (CAR-T) therapy. CAR-T therapy is a type of immunotherapy that genetically modifies the patient's own T cells to target and kill cancer cells. [12, 13] In MM patients specifically, B cell maturation antigen (BCMA) targeted CAR-T therapy has been highly successful in newly diagnosed and relapsed MM patients. [13] Furthermore, clinical studies have found that CAR-T therapy has a 76% remission rate and conclude better 5-year patient outcomes.[14] Today there are two FDA-approved CART-T therapies for MM, Carvykti and Abecma, with others in development.[15] Despite successes, there are notable challenges with CAR-T therapy including cytokine release syndrome (CRS) and neurotoxicity, which may be managed with appropriate and timely interventions. [13] CAR-T therapy is also costly and requires specialized manufacturing facilities, limiting its availability to patients. Importantly, new treatments outside of CAR-T therapy are also in development with novel targets, namely Selinexor. Selinexor targets Chromosome Region Maintenance 1 (XP01), a transporter of nuclear proteins with a key role in cell cycle regulation and proliferation.[16] XP01 has been implicated in several solid tumors, such as lung cancer, and is overly expressed in MM. Overexpression of XP01 allows tumor suppressor proteins, such as Rb, p53, and p21, to be exported into the cytoplasm rendering them inactive and therefore increasing anti-apoptotic cell signaling.[16, 17] Selinexor covalently binds to the “cargo-binding” groove of XP01 which prevents the displacement of tumor suppressor proteins mentioned above.[17, 18] As a result of its pre-clinical and clinical trial success, the FDA approved Selinexor as a combination therapy with dexamethasone for patients with more than one prior therapy and at least 4 prior dexamethasone therapies.[19, 20] While MM continues to be a challenging cancer to treat, therapies such as CAR-T therapy and Selinexor provide MM patients with new hope for lasting remission. New clinical trials, such as NCT05177536 to test iberdomide maintenance therapy, are underway to explore the efficacy of new therapies and interventions to improve MM outcomes.
Benefits of Immuno-Oncology for Colorectal Cancer
Colorectal cancer (CRC) is a leading cause of cancer deaths globally. Advances in early detection have improved survival rates, but patients diagnosed with metastatic CRC still have stubbornly poor 5-year survival rates. Standard treatments for CRC include surgery, chemotherapy, and radiotherapy, but alternative immuno-oncology therapies are showing promising results in CRC patients. Here we highlight advances in immuno-oncology therapies that are being used to treat CRC patients or are being pursued in preclinical and clinical studies. CRC Subtypes Molecular genetic analysis of CRC tumors is a critical diagnostic tool for classifying and treating this cancer. CRC tumors are typically classified as mismatch repair-deficient/microsatellite instability-high (dMMR—MSI-H) tumors, which have a high overall mutation burden, or mismatch repair-proficient/microsatellite instability-low (pMMR—MSI-L) tumors, which have a lower mutation burden[1]. Defects in MMR are associated with the accumulation of mutations and are caused by defects in mismatch repair proteins, and these defects are typically detected by frame-shift mutations in DNA repeat regions known as microsatellites. Mutations in the BRAF oncogene, particularly the activating V600E mutation, comprise a distinct subset of CRC. BRAF is a component of the mitogen-activated protein kinase (MAPK) pathway that normally functions downstream of the epidermal growth factor receptor to regulate transcription of genes involved in cellular growth and survival, but the BRAF-V600E mutation results in constitutive activation of BRAF and uncontrolled cellular proliferation and tumor growth[2]. Immuno-Oncology Interventions Immuno-oncology approaches that target immune checkpoint blockade have proven effective for several cancers, including CRC. T cells can initiate effective anti-tumor responses under ideal conditions, but the immunosuppressive tumor microenvironment of some cancer types inhibits T cell activation, usually through engagement of immune checkpoint molecules like PD-1 and CTLA4. For more than a decade, the development and use of immune checkpoint inhibitors (ICIs) has transformed the treatment of melanoma[3,4] and non-small cell lung cancer[5,6]. The first FDA-approved treatments include a monoclonal antibody (mAb) that targets CTLA4 (ipilimumab) and two mAbs that target PD-1 (pembrolizumab and nivolumab). These early studies suggested that tumors with high mutation burdens respond well to ICI, which may be due in part to the generation and presentation of tumor neoantigens that are recognized as non-self and can be targeted by cytotoxic T cells[7]. CRC tumors with the dMMR—MSI-H signature have a high mutational burden and typically have a high level of CD4+ and CD8+ tumor-infiltrating lymphocytes (TILs). These cells have shown elevated expression of PD-1, PD-L1, and CTLA4, which suggests that they may respond well to ICIs[8]. Indeed, several recent clinical studies have shown improvements in overall survival and progression-free survival in patients treated with individual or combined ICIs that target PD-1. Of note, interim results from a phase II trial that enrolls patients with MMRd locally advanced rectal cancer to receive neoadjuvant dostarlimab, a PD-1 inhibitor, showed sustained complete response 1 year after the end of treatment in all patients[9]. Other current studies are evaluating next-generation PD-1 inhibitors combined with chemotherapy and/or other biologics such as an anti-VEGF and anti-EGFR mAb[10]. In contrast, patients with pMMR–MSI-L have shown poor responses to PD-1 or CTLA4 blockade alone or in combination, which has led to the development of trials that explore different treatment combinations, including inhibitors of the MAPK pathway or angiogenesis[11]. Promising results have been presented in a phase I/II clinical trial (NCT04017650) in which patients with MSI-L and BRAFV600E were treated with BRAF+EGFR inhibitors, to induce a transient MSI-H phenotype, plus anti-PD-1 antibody[12]. Next-Generation Treatments Several cutting-edge immuno-oncology therapies are being explored for the treatment of CRC. Beyond combinations of individual antibodies, researchers are engineering bispecific antibodies that bind to tumor cells and T cells simultaneously to enhance anti-tumor T cell responses. One such bispecific antibody, CEA-TCB, is being tested in phase I trials alone or in combination with anti-PD-L1 to treat metastatic CRC[13]. Another novel approach includes adoptive cell therapies like chimeric antigen receptor (CAR) T cells, which are T cells collected from the tumor tissue or peripheral blood of a patient and engineered to bind to tumor antigens and potentiate anti-tumor responses. Oncolytic virus and bacteria-based vaccines are also being studied as potential CRC treatments. Besides treatments designed on the tumor's intrinsic genetic background, much effort is being put into deciphering the tumor microenvironment, with a particular focus on its interplay with the gut microbiota to modulate inflammation and immune response, known to be involved in the metastatic onset. These studies are leading to the identification of inflammatory/immune signatures that inform the therapeutic agents to be used to target the pro-oncogenic microenvironment and reactivate the immune system[14]. The advancement of immuno-oncology is already transforming the treatment of CRC and will contribute to better outcomes for all types of CRC in the decades to come.
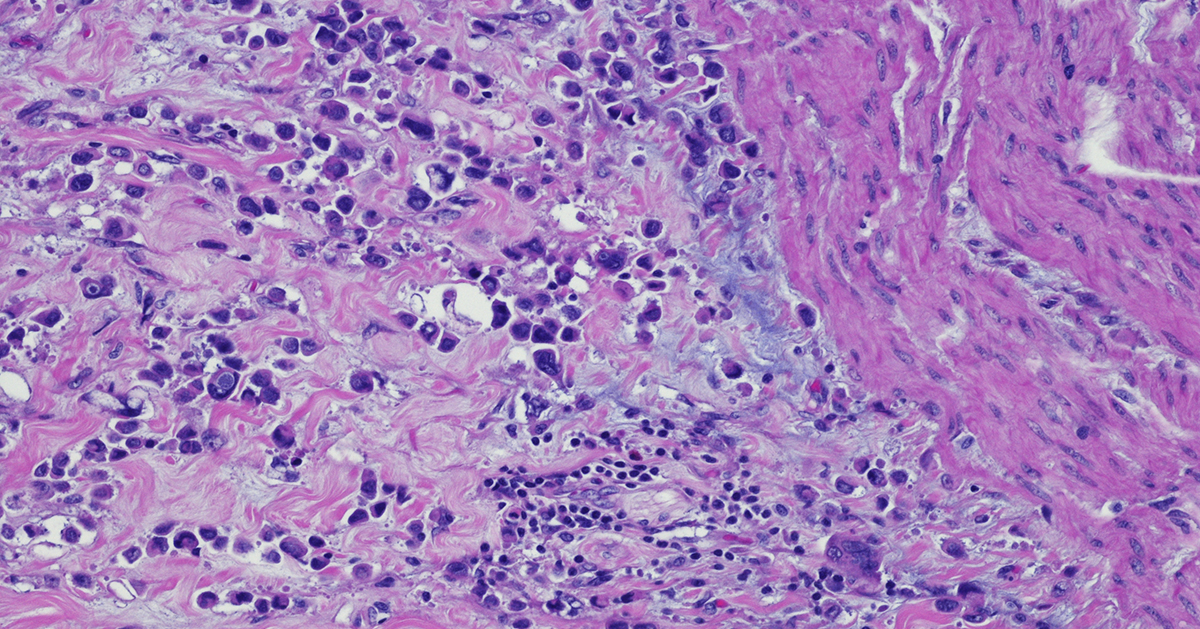
Immunohistochemistry: a Powerful Tool in Cancer Research
Immunohistochemistry (IHC) is a technique that originates in the early twentieth century but continues to be a valuable method that forms the backbone of molecular pathology. IHC is used for histological examination of tissues and specifically detects the presence of a molecule, such as a tumor antigen. IHC uses antibody-based labeling in which the primary antibody detects the target of interest and the secondary antibody detects the primary antibody which is linked to a molecule for microscopic visualization. Many different secondary antibody labeling modalities exist, including fluorescence, enzyme-mediated reactions and colloidal gold, and different labels are suited to specific microscopy platforms. Consider these five aspects of IHC as you implement this technique in preclinical cancer research: Quantitative measurements. IHC can be used as a qualitative measurement, but unlike many other visualization techniques, IHC can also be used as a quantitative measurement because antibodies that label specific parts of tissues or cells can be counted by a pathologist or with a computer-aided system. Developing robust validated quantitative IHC staining and visualization methods allows researchers to rely on the accuracy of this data. Customizable. IHC methods can be adapted to detect any cellular marker, given that a monoclonal antibody exists or can be made that specifically detects this marker. Validation of a new primary or secondary antibody also includes determining any off-target staining caused by these antibodies as this can be a critical determinant in the utility of an antibody. The ability to customize IHC in this manner is crucial to preclinical research that seeks to identify new biomarkers associated with tumor progression or immunotherapy efficacy. Flexibility. IHC can be used on almost any tissue type so long as it is processed correctly. Tissue samples from model animals as well as clinical biopsies can be fixed and sectioned in advance and stained at later times. Tumor microarrays (TMAs) can also be created and stained for evaluation of novel tumor markers or screening efficacy of drug candidates. This flexibility in sample type and handling highlights the overall utility of IHC. Automation. Currently, several different systems exist for automated IHC staining, and advances in digital analysis of IHC samples have allowed larger batches of clinical samples to be processed and evaluated. Comparison of automated IHC methods with manual methods has determined that these new approaches are accurate, sensitive, and reproducible. The big picture. Several methods exist for staining different targets in an IHC sample, and this allows scientists and clinicians to gain critical insights into which cells and molecules are present in the tumor microenvironment (TME), including levels of immune checkpoint molecules or infiltration of critical anti-tumor cell types. Consider how IHC can complement other techniques that look at the tumor microenvironment, including flow cytometry and RNAseq. IHC will continue to be a powerful tool in preclinical and clinical cancer research. Consider revisiting this classic technique as it has matured with the technological advances of the 21st century. Champions Oncology’s histology and immunohistochemistry services are custom developed and fully optimized to meet your needs in preclinical research. With industry leading pathology expertise and innovative automated technology, Champions provides you with the highest quality endpoints for your in vivo and ex vivo studies.

Uncovering Advantages and Disadvantages of Ex Vivo Culture in Preclinical Cancer Research
Ex vivo culture systems have been instrumental to preclinical oncology research because they enable researchers to study basic features of tumor cells and carry out large-scale screens of drugs or biologics. Ex vivo cultures do not fully recapitulate physiological conditions, although some advances have been made by developing three-dimensional (3D) culture methods. Here we highlight the advantages and disadvantages of using ex vivo cultures for preclinical oncology research. Cell culture: simple, inexpensive, but imperfect Ex vivo cell cultures of tumors were first developed as adherent two-dimensional (2D) monolayers that were grown in culture flasks or flat dishes. This approach is useful for studying tumors' basic cell biology and carrying out drug screens and preliminary toxicology and pharmacokinetic studies[1]. 2D cultures can be developed rapidly and cultured over longer periods of time using readily available tissue culture reagents. Unfortunately, 2D culture systems are limited in their applications because they do not accurately model the tumor microenvironment, and tumor cell morphology and function change under these culture conditions. Over the last several decades, 3D cultures have advanced significantly[2]. Several different systems now exist for 3D cultures, including liquid suspension cultures passaged on non-adherent plates, cultures grown in semi-solid substrates like soft agar or Matrigel, and cells grown on scaffolds like collagen. 3D culture systems better mimic characteristics of the tumor microenvironment, including cell-cell interactions, hypoxia, and reduced sensitivity to drug treatment[3]. (Visit our dedicated webpage to learn more about Champions' 3D tumor model offer) 3D cultures for drug discovery have become widely adopted over 2D models because they better predict in vivo efficacy and metabolic responses to drug treatments. A major advance in 3D tumor culture systems is the development of 3D spheroid and organoid cultures[4]. Spheroids are clusters of cells derived from tumors or other complex tissues and they cluster together through cell-cell adhesion. Organoids are more complex cell clusters derived from stem cells and can self-assemble and regenerate to form a smaller version of the original tumor or organ tissue. Tumor spheroids are widely used for drug screening and cytotoxicity assays and can be formed from dissociated tumor tissue or circulating tumor cells[5]. (Read our blog "Using 3D Ex Vivo Tumor Models for Oncology Research: An Expert Guide") Tumor organoids are even more accurate reflections of tumors growing under physiological conditions and are now widely used to compare the efficacy of standard-of-care treatments versus targeted therapies in addition to their use in drug screens[6]. Spheroids and organoids can undergo genomic editing as well, and this approach has been instrumental in identifying mutations that drive tumor growth or cause drug resistance[7-8]. (Read our blog "A Step-by-Step Guide to Design a 3D Tumor Model Co-Culture Study") Ex vivo + in vivo = the most effective strategy Animal models, especially patient-derived xenograft mouse models, are a critical bridge between preclinical studies and clinical trials. However, due to the lower costs and the compatibility with high throughput analysis, ex vivo tumor cultures are still the preferred method for ranking therapeutic agents, determining drug combination regimens, investigating mechanisms of action, and validating targets. Additionally, ex vivo tumor cultures are essential when studying certain hematological tumors, which are difficult to engraft and cannot be passaged in vivo because of their “liquid” nature. Both ex vivo and in vivo studies continue to work together to advance oncology research, but each year brings new advances in ex vivo culture systems that enhance their overall impact in predicting treatment responses.