
Trends in Oncology
A KRAS mutation is not a strategy: Why allele, co-mutation, and tumor type matter
In our last blog post, we explored why resistance has become the defining challenge in KRAS drug development. As the field has matured, it has become clear that early activity alone is not enough, and understanding why tumors respond or not, and how they escape treatment is now essential. It also raises the deeper issue of if response and resistance vary so widely, then a critical assumption at the heart of many programs needs to be challenged: Can “KRAS-mutant” still be treated as a single category in drug development? The evidence increasingly suggests the answer is no. The problem with treating KRAS as one market As KRAS-targeted therapies have moved forward, many development strategies have been framed broadly around “KRAS-mutant tumors.” On the surface, this makes sense as KRAS is one of the most common oncogenic drivers across multiple cancer types. However, in practice, this simplification hides important biological differences. Across KRAS-mutant tumors, responses to targeted therapies are highly variable, with some settings showing meaningful activity, while others demonstrating limited or short-lived responses. Even within the same tumor type, outcomes can differ significantly across patients. This variability is not random and reflects the underlying biological context, which now matters more than ever. Allele matters more than ever Not all KRAS mutations are the same, with variants such as G12C, G12D, and G12V differing in their biochemical properties, downstream signaling behavior, and sensitivity to targeted inhibition. As more therapies are developed beyond the initial wave of KRAS G12C inhibitors, these differences are becoming increasingly relevant. Emerging preclinical data shows that response patterns can vary across KRAS allele types, influencing both the depth and durability of response. For drug developers, this means that evaluating an asset against a generic “KRAS-mutant” panel is no longer sufficient, and the real question to now ask is: Which KRAS variants does your therapy work against, and under what conditions? Co-mutations shape response and resistance KRAS does not operate in isolation. Tumors often carry additional genetic alterations that influence pathway signaling, tumor behavior, and therapeutic response. These co-driver mutations can enhance sensitivity in some contexts or drive resistance in others. Across clinically relevant models, differences in co-mutation profiles have been linked to distinct response patterns and resistance mechanisms, which has two important implications: A therapy that appears broadly effective in a simplified model system may fail in more complex, clinically representative settings. Co-mutation context can represent an opportunity. Understanding these relationships can help identify patient subsets that are more likely to benefit or reveal combination strategies that overcome resistance. Tumor type defines the biological landscape KRAS mutations occur across multiple tumor types, including non-small cell lung cancer, colorectal cancer, and pancreatic ductal adenocarcinoma. Each tumor type exists within its own biological environment, shaped by factors such as tissue origin, microenvironment, and lineage-specific signaling networks. These differences influence how KRAS-driven pathways behave and how tumors respond to targeted therapies. As a result, a therapy that performs well in one indication may not translate directly to another. This is why preclinical strategies must be designed with indication in mind. Testing a KRAS program in the wrong context can lead to misleading conclusions about its potential. Why context-aware model selection is critical Taken together, these factors make one thing clear: Context is a requirement, rather than a refinement. Effective KRAS studies must move beyond generic model selection and instead reflect the biological diversity seen in the clinic, which includes: Selecting models that represent specific KRAS variants Capturing diverse co-mutation landscapes Matching studies to relevant tumor types Incorporating clinically meaningful patient characteristics Modern preclinical platforms are increasingly designed to meet these needs by combining deeply characterized patient-derived models with molecular and clinical annotation. This approach allows teams to design studies that are not only more realistic, but also more informative. Better context leads to better decisions When KRAS programs are evaluated within the right biological context, the quality of decision-making improves significantly, allowing teams to: Rank assets more accurately Identify the most promising indications Develop more precise biomarker strategies Design more rational clinical trials Perhaps, most importantly, they can avoid overinterpreting results that may not translate outside of a narrow experimental setting. In a competitive and rapidly evolving field, this clarity can make the difference between advancing the right candidate and pursuing a path that ultimately fails in the clinic. From resistance to real-world biology In our previous article, we highlighted the importance of modeling resistance to understand how tumors respond over time, and in this post we have expanded on that idea. Resistance does not emerge in a vacuum. It is shaped by the biological context of the tumor, including allele, co-mutation, and tumor type. Ignoring that context risks misunderstanding both response and resistance. What comes next If biological context shapes response, then treatment history is another critical factor that must be considered. In reality, most patients entering clinical trials are not treatment-naive. Their tumors have been shaped by prior therapies, and those therapies influence how they respond to potential new agents. In our next post in this series, we will explore why treatment-naive models are increasingly insufficient and why pretreated, clinically aligned models are essential for studying KRAS programs in today’s landscape. Talk to us If you are developing a KRAS-targeted therapy, understanding how allele, co-mutation, and tumor type shape response is no longer optional. Talk to our team about designing context-aware preclinical studies that generate data you can trust and act on. Have a Question?

The next KRAS battleground: modelling resistance before the clinic
In our previous posts, we explored how KRAS has entered a new era and why benchmarking has become essential for differentiating assets in an increasingly competitive landscape. But as the field evolves, a new reality is becoming clear: In KRAS drug development, early activity is no longer enough. The programs that succeed will be the ones that understand resistance before they reach the clinic. The shift from proof of concept to durability For decades, KRAS was defined by a single challenge: proving that it could be drugged at all, a barrier that has now been crossed. With multiple KRAS and pan-RAS targeted therapies advancing through the pipeline, the question is no longer whether a compound can generate tumor shrinkage, it is whether that response will be deep, durable, and reproducible across patient populations, and is where many programs begin to diverge. Preclinical studies often still focus on early efficacy signals and in reality, clinical success depends on something much more complex: how tumors respond over time and how quickly they adapt. Why resistance is now the defining challenge in KRAS Recent preclinical work in KRAS-mutant models shows that responses to targeted therapies are highly variable, with clear differences in both intensity and duration. Some tumors fail to respond at all, with others responding initially and then relapsing as resistant clones emerge. Understanding these patterns is now critical to making informed development decisions. Across diverse KRAS-mutant settings, response heterogeneity has been linked to KRAS allele type, co-driver mutations and prior treatment exposure. These variables reflect the biological diversity that defines real patient populations and ignoring them in preclinical work means overlooking the very factors that determine clinical outcomes. Not all resistance is the same To model resistance effectively, it is important to recognize that there are two distinct challenges: 1. Intrinsic resistance: Some tumors show little or no response to treatment from the outset. These cases often reflect underlying biology that makes the therapy ineffective in that context. 2. Acquired resistance: Other tumors respond initially, but over time develop mechanisms that allow them to escape treatment pressure. Advanced KRAS studies now capture both dynamics by: Comparing responders versus non-responders at baseline Tracking tumors longitudinally after initial regression Analyzing the molecular changes that drive resistance over time Without this dual perspective, programs risk misunderstanding both the limits and the true potential of their assets. Why traditional preclinical models fall short So, if you know that resistance is the defining challenge, why do so many preclinical studies fail to capture it? The answer usually lies in how those studies are designed. Many studies designs still rely on small numbers of models, treatment-naive systems and single endpoint efficacy readouts. These approaches can identify whether an asset has activity. They struggle to answer more important questions, like: Which patients are likely to respond? How durable are those responses? What mechanisms will drive relapse? Clinically relevant preclinical platforms are moving toward combining diverse patient-derived models, treatment history, and deep molecular characterization to better reflect real-world tumor biology. This shift now makes it possible to evaluate how therapies perform beyond the initial response. What traditional KRAS studies miss (side by side comparison) Traditional studies Resistance-aware studies Few models Diverse cohorts Treatment-naive Pretreated Single endpoint Longitudinal Limited readouts Multi-omic What better KRAS studies should capture It is clear, to move from proof of concept to true translational insight, KRAS studies need to evolve. At a minimum, that means incorporating: Biological diversity across KRAS variants and tumor types Co-mutation context to reflect real genetic drivers Treatment history to model clinically relevant resistance Longitudinal sampling to track response and relapse Multi-omic profiling to uncover mechanisms of sensitivity and escape These elements allow teams to move beyond simple efficacy metrics and begin to answer the questions that matter most for clinical success. Why resistance modeling changes decision making When resistance is built into preclinical strategy, the impact directly informs things like; Asset ranking between competing KRAS programs, Indication selection across tumor types; Biomarker strategy for patient segmentation, and; Combination approaches designed to prevent or overcome resistance. Resistance modeling is not just about understanding failure. It is about designing for success earlier in development. From benchmarking to biology Our previous post highlighted why benchmarking is now essential in KRAS drug development. However, benchmarking alone is not enough. To generate meaningful comparisons, studies must account for the biological factors that drive response and resistance. Without that context, even rigorous comparisons can lead to misleading conclusions. What comes next If resistance varies so widely across KRAS-mutant tumors, it raises an important question: Can “KRAS-mutant” still be treated as a single category in drug development? In our next blog post, we explore why the answer is no and why allele, co-mutation, and tumor type must shape every aspect of KRAS strategy. If you are advancing a KRAS program, designing studies that capture both response and resistance is no longer optional. Talk to our team about building preclinical strategies that reflect real patient biology and generate data you can act on. Have a Question?

Why Benchmarking is Now Essential in KRAS Drug Development
The KRAS landscape has changed. As next-generation inhibitors continue to demonstrate clinical impact, the expectations placed on preclinical programmes are increasing. At the centre of this shift is a simple but critical concept. Benchmarking is no longer a differentiator, but a requirement. The Rise of a Clinical Reference Point As therapies such as daraxonrasib advance through clinical development, they establish a new baseline for efficacy. This baseline becomes the reference against which all new KRAS-targeting programmes are evaluated. For preclinical scientists, this fundamentally changes the design of studies. It is no longer sufficient to show that a compound reduces tumor growth in a subset of models. The real question is whether that activity represents meaningful improvement over what is already achievable. Without benchmarking, this question cannot be answered. What Benchmarking Enables Benchmarking provides context and transforms isolated, siloed data into translatable data; the basis on which your decision-making can be trusted. When preclinical results are generated against a known standard, researchers can: Quantify relative efficacy Identify tumor subtypes where a therapy may outperform existing options Understand where resistance patterns differ Evaluate the potential for combination strategies In KRAS-mutant tumors, where heterogeneity is high and resistance is common, this context is essential for making informed decisions. The Importance of Scale and Annotation Not all benchmarking datasets are equal. To be meaningful, benchmarking must be built on three core components: 1. A sufficiently large and diverse model set: KRAS mutations span multiple tumor types and molecular contexts, and a robust dataset must reflect this diversity. 2. Deep molecular characterisation: Genomics and transcriptomics alone are not enough to fully capture tumor biology. Protein-level and pathway-level data provide additional resolution that is critical for understanding response. 3. Clinical relevance: Models must reflect real patient biology, including treatment history and resistance mechanisms. At Champions Oncology, we have generated daraxonrasib response data across more than 50 KRAS-mutant PDX models spanning lung, colorectal, and pancreatic cancers. Each model is fully annotated with clinical and molecular data, enabling direct linkage between response and biology. This is not a conventional screening dataset but a translational framework enabling decision-making. Moving Beyond Binary Outcomes Traditional preclinical studies often present results in binary terms. Tumors respond or they do not. While this can be useful at an early stage, it lacks the resolution needed for modern drug development. Benchmarking enables a more nuanced view and instead of asking whether a tumor responds, scientists can ask: How does this response compare to existing therapies? What molecular features distinguish responders from non-responders? Which resistance pathways are likely to emerge? This level of analysis is essential for designing therapies that succeed in the clinic. Implications for Clinical Strategy The value of benchmarking extends beyond the preclinical stage and directly informs clinical development. By linking response data to molecular features, benchmarking datasets can support: Biomarker-driven patient selection Rational combination therapy design Selection of optimal indications Identification of expansion cohorts In a competitive KRAS landscape, these insights provide a critical advantage. A New Standard for Preclinical Research As the field continues to evolve, benchmarking will become a standard component of preclinical workflows. Teams that fail to incorporate benchmarking risk generating data that lacks context and relevance. The future of KRAS research lies in integrated, data-rich approaches that connect biology, pharmacology, and clinical strategy. Benchmarking is the foundation of that future. What Comes Next In the next blog in this series, we examine why many KRAS preclinical models fail to translate into clinical success and how integrated, multi-omic approaches are addressing this gap. Continue exploring KRAS blog posts

KRAS Has Entered a New Era. Why Your Preclinical Strategy Must Change Now
KRAS has long been one of the most important targets in oncology, and for decades it was widely considered “undruggable”. Fortunately, that perception has changed rapidly and today, KRAS-targeted therapies are no longer theoretical. They are clinically validated, increasingly effective, and reshaping how drug development programmes are designed. The transition from early KRAS inhibitors (KRASi) to next-generation compounds represents a fundamental shift in how our field approaches targeted therapy. For preclinical and translational teams, this shift introduces both opportunity and risk. Programs that adapt quickly will be well positioned to succeed, whereas those that rely on outdated models and assumptions may fall behind. A New Standard of Care Is Emerging Recent clinical data for pan-RAS(ON) inhibitors, such as daraxonrasib, signal a turning point in KRAS drug development. As these therapies move toward becoming standard of care, they establish a new benchmark against which all future KRAS-directed agents will be measured. This has immediate implications for preclinical strategies, and it is no longer sufficient to demonstrate activity in isolation. Every new therapy must now be evaluated relative to an evolving clinical reference point. For companies developing therapies in KRAS-mutant indications such as non-small cell lung cancer (NSCLC), colorectal cancer, and pancreatic cancer, the key question has changed. It is no longer “Does this work?” but “Does this outperform or complement what already exists?”. Why Traditional Preclinical Approaches Are No Longer Enough Historically, KRAS drug development has relied heavily on simplified systems such as cell lines or limited in vivo studies. While these approaches played an important role in early discovery, they are insufficient for today’s demands. KRAS biology is highly context dependent and mutation status alone does not determine response. Co-occurring genomic alterations, transcriptional programmes, and protein-level signalling all contribute to therapeutic sensitivity and resistance. As a result, preclinical systems that lack clinical relevance or molecular depth fail to capture the complexity of real tumours. This creates a disconnect between preclinical findings and clinical outcomes, increasing the risk of late-stage failure. The Role of Clinically Relevant Models To address this challenge, there is a growing shift toward clinically relevant models such as patient-derived xenografts (PDXs) with integrated molecular characterisation. These models enable a deeper understanding of tumor biology and provide a more accurate representation of patient response. Importantly, when these models are combined with longitudinal data and treatment history, they allow researchers to study both intrinsic sensitivity and acquired resistance. This is essential in KRAS, where resistance is not an exception but an expected outcome. Benchmarking as the New Foundation In this evolving landscape, benchmarking is becoming the basis of an effective preclinical strategy, and access to datasets that include response to current or emerging standards of care allow scientists to: Identify responder and non-responder populations Understand the molecular drivers of response Design rational combination strategies Prioritise assets with the highest likelihood of success At Champions Oncology, we recognised that benchmarking would become essential in the KRAS space. Our work has focused on building clinically annotated datasets that connect tumor response directly to molecular biology. This enables a level of insight that extends far beyond traditional screening approaches. From Activity to Decision-Making The ultimate goal of preclinical research is not only to generate data, but to inform your decision making. In KRAS drug development, the stakes are high. Clinical trials are expensive, timelines are long, and the competitive landscape is rapidly evolving. Preclinical data must therefore do more than demonstrate activity. It must guide: Patient selection strategies Combination therapy design Clinical trial structure Pipeline prioritisation This requires a shift from descriptive models to more predictive, translational systems. Looking Ahead KRAS has entered a new era. The science is advancing, the clinical landscape is evolving, and expectations are rising across the industry. For preclinical teams, this is a moment to reassess how studies are designed, how models are selected, and how data are interpreted. The organisations that lead in this space will be those that align their strategy with the realities of the current landscape, not the assumptions of the past. In the next blog in this series, we explore why benchmarking is no longer optional in KRAS research and how it is reshaping preclinical decision-making. Poster: Molecular determinants of sensitivity and resistance to the pan-RAS(ON) inhibitor daraxonrasib
Daraxonrasib Is Rewriting the Standard of Care. Here's Why Preclinical Benchmarking Matters Now
A Standing Ovation and a New Standard of Care At the 2026 ASCO Annual Meeting, the RASolute 302 Phase 3 trial delivered what many are calling the most significant advance in pancreatic cancer treatment in decades. Daraxonrasib, a first-in-class oral RAS(ON) multi-selective inhibitor developed by Revolution Medicines, nearly doubled overall survival (OS) in previously treated metastatic pancreatic ductal adenocarcinoma (PDAC), achieving a median OS of 13.2 months compared to 6.7 months with standard chemotherapy (HR 0.40; p<0.0001). Progression-free survival followed the same trajectory: 7.3 months versus 3.5 months. The results, published simultaneously in The New England Journal of Medicine, drew a standing ovation during the plenary session. ASCO's chief medical officer called it "a grand slam," and the ASCO-selected commentator described starting to cry in clinic after seeing the data. This is no longer a drug to watch. Daraxonrasib is poised to become the new standard of care for second-line metastatic PDAC, with first-line combination trials already underway. For drug developers working in KRAS-driven cancers, the question is no longer whether daraxonrasib will reshape the treatment landscape, but how fast, and whether your preclinical program is ready. Why Benchmarking Against Daraxonrasib Is Now Essential More than 90% of pancreatic cancers harbor KRAS mutations, making it one of the most RAS-addicted tumor types in oncology. As daraxonrasib transitions from investigational therapy to standard of care, it will become the benchmark against which new agents, novel combinations, and next-generation therapies are measured. This shift has immediate implications for preclinical strategy. Scientists developing therapies in KRAS-mutant NSCLC, colorectal cancer, and PDAC need access to clinically annotated models with existing daraxonrasib response data to design meaningful benchmarking and combination studies. Without this data, preclinical programs risk testing against an outdated treatment landscape. Yet despite the urgency, a significant gap exists. Most contract research organizations and preclinical providers do not have daraxonrasib response data, the KRAS-mutant patient-derived xenograft (PDX) models needed to generate it, or the multi-omic depth required to interpret results in a translational context. Champions Oncology Has Already Built the Dataset At Champions Oncology, we recognized early that daraxonrasib would become a defining compound in the KRAS space. That is why we have already screened over 50 KRAS-mutant PDX models across NSCLC, colorectal cancer, and PDAC, generating a unique and comprehensive daraxonrasib benchmarking dataset that is available today. Figure 1: Tumor response and genomic landscape of KRAS-mutant PDX models treated with Daraxonrasib (RMC-6236) This is not a standard drug screen. Every model in the cohort is fully annotated with clinical history, treatment status, and deep molecular profiling, including whole exome sequencing, RNA sequencing, and integrated genomic analysis. The resulting waterfall plots and genomic annotation panels allow scientists to rapidly identify responder and non-responder models matched to their therapeutic hypothesis, and to understand the molecular context driving each outcome. Understanding what drives resistance. Differential gene expression analysis of responding versus non-responding KRAS G12D-mutant PDAC models has identified transcriptional programs associated with primary resistance to daraxonrasib, providing actionable insights for biomarker development and patient selection strategies. Modeling acquired resistance in real time. Because secondary resistance is an inevitable feature of targeted therapy, we have developed acquired resistance models under continuous daraxonrasib treatment pressure. Models CTG-2473 (pancreatic, KRAS G12D) and CTG-0068 (colorectal, KRAS G12D) both demonstrated initial tumor regression followed by regrowth, recapitulating the resistance patterns expected in patients. These models create a powerful translational framework for testing next-line agents, rational combination strategies, and treatment sequencing approaches. Going deeper with multi-omic prediction. Champions' Pharmaco-Pheno-Multiomic (PPMO) integration platform takes this further by layering whole-cell proteomics, cell surface proteomics, genomics, and transcriptomics across 56 KRAS-mutant PDX models treated with daraxonrasib. The resulting computational model achieves 80% prediction accuracy for drug response, substantially outperforming RNA-only approaches, which plateau at approximately 60%. This level of biological resolution is critical for identifying the molecular determinants of sensitivity and resistance that standard biomarker panels miss. Download the Full Poster These findings were first presented at AACR 2026 (Poster 1884): Molecular Determinants of Sensitivity and Resistance to the Pan-RAS(ON) Inhibitor Daraxonrasib (RMC-6236) Across KRAS-Mutant Patient-Derived Models. For the complete dataset, including response profiles, genomic annotations, and resistance modeling data, download the full poster here. For additional context, read our companion blog: Understanding Sensitivity and Resistance to Pan-RAS(ON) Inhibition Across KRAS-Mutant Tumors. The Landscape Is Shifting. Let's Talk About Your Program. Daraxonrasib is changing how KRAS-targeted therapies will be developed and evaluated. Whether you are benchmarking a novel compound against an emerging standard of care, designing combination strategies, or investigating mechanisms of primary and acquired resistance, we have the models, the data, and the translational depth to support your program. Contact us today to discuss your study
Clinically Relevant 3D Tumor Models for HER2-Targeted ADC Development
Expanding the Preclinical Toolbox with a Novel Patient-Derived Organoid Platform The human epidermal growth factor receptor 2 (HER2) plays a central role in regulating cell growth, division, and survival. When overexpressed, it acts as an "on switch", driving accelerated cell growth and uncontrolled tumor spread. This biology underlies the aggressive behavior observed in HER2-positive cancers, which is associated with higher recurrence rates and poorer patient outcomes. While genomic changes in HER2 are most frequently discussed in the context of breast cancer, HER2 alterations are also found in non-small cell lung, ovarian, colorectal, and pancreatic cancers. HER2-Targeted ADCs in Cancer Therapy HER2-targeted therapies have significantly changed the treatment landscape and brought new hope and better outcomes for patients. Among them, HER2-targeted antibody-drug conjugates (ADCs) have emerged as a particularly promising approach, combining the specificity of antibodies with the potency of cytotoxic payloads. These agents enable targeted delivery directly to HER2-expressing tumor cells while limiting off-target toxicity. FDA approved drugs in this class include trastuzumab emtansine (T-DM1, Kadcyla®) and trastuzumab deruxtecan (T-DXd, Enhertu®), both of which have demonstrated meaningful clinical responses (Figure 1). A growing number of next-generation HER2 ADCs are now in development. Figure 1. FDA approved HER2-targeted ADCs trastuzumab emtansine (T-DM1, Kadcyla®) and trastuzumab deruxtecan (T-DXd, Enhertu®). Figure adapted from Joubert et al1. As ADCs become an increasingly important therapeutic modality, the limitations of conventional preclinical models have become magnified. Drug developers need better, more physiologically relevant and predictive preclinical models to support ADC development, ones capable of reproducing tumor architecture, heterogeneity, and treatment response. Limitations of Traditional 2D Preclinical Models for ADC Evaluation Despite their widespread use, traditional 2D cell culture models have well-recognized drawbacks with respect to evaluating targeted therapies. They often fail to capture key biological features of tumors and inadequately replicate the complex tumor microenvironment (TME) that influences drug response in patients. Target accessibility, receptor density, internalization kinetics, and payload penetration are all influenced by three-dimensional tumor structure, features which are largely absent in 2D cultures. As a result, 2D assays may overestimate drug potency or fail to distinguish on-target activity from nonspecific cytotoxicity, reducing their predictive value for clinical translation. In contrast, growing tumor cells as 3D organoids allows for a spatially organized structure that more closely resembles the in vivo microenvironment and architecture of patient tumors. Champions has developed a scalable 3D screening platform based on HER2-positive patient-derived xenograft organoids (PDXOs). Derived from well-characterized PDX models, our ex vivo 3D organoids maintain clinically translatable HER2 expression, providing a relevant and predictive platform for evaluating the efficacy of HER2-targeted ADCs. Validation of HER2-Positive PDXO Models HER2-positive ex vivo PDXO breast cancer models generated from our TumorGraft3D (CTG-3D) platform were evaluated for HER2 expression as well as functional responses to the clinically approved HER2-targeted ADCs, trastuzumab emtansine (T-DM1) and trastuzumab deruxtecan (T-Dxd). The workflow is shown in Figure 2. Figure 2. Workflow overview to evaluate HER2-targeted ADCs using breast cancer PDXO models. HER2 Expression Immunohistochemical (IHC) analysis of PDXO tissue blocks demonstrated complete and intense membrane staining (HER2 score 3+) across HER2-positive models. Consistent with these findings, flow cytometric analysis of enzyme-dissociated organoids identified HER2-positivity in 61.8% to 90.1% of cells. In contrast, the HER2-negative control PDXO model showed no detectable HER2 staining by IHC and negligible HER2 expression by flow cytometry, confirming assay specificity and the absence of nonspecific background signal. A subset of the results is shown in Figure 3. Figure 3. HER2 expression assessed by IHC and flow cytometry analysis. A) HER2-positive PDXO and B) HER2-negative PDXO model. Together, the IHC staining and flow cytometry results are consistent with the clinical annotation and indicate that HER2 expression is maintained relatively homogeneously in breast cancer PDXO models. Response to HER2-Targeted ADCs After confirmation of HER2 expression, the PDXO models were evaluated for their response to two clinically approved HER2-targeted ADCs, trastuzumab deruxtecan (T-DXd) and trastuzumab emtansine (T-DM1). Both ADCs were tested against HER2-positive and HER2-negative breast cancer PDXOs and reference breast cancer cell lines. In addition to intact ADCs, corresponding cytotoxic payloads (Exatecan and DM1), naked antibody (trastuzumab), and—for T-DXd—an isotype-payload conjugate (IgG-DXd) were evaluated to distinguish potency, selectivity, and target-dependent activity. Drug response was measured using the CellTiter-Glo® viability assay following six days of incubation with the ADCs and control treatments. Figure 4. Representative dose-response curves (IC₅₀). A) HER2-positive PDXO model comparing exatecan (payload) and trastuzumab deruxtecan (T-DXd) and B) control naked antibody trastuzumab and isotype-payload conjugate IgG-DXd. Across HER2-positive breast cancer PDXO models and control cell lines, ADCs demonstrated greater efficacy and specificity compared with naked antibody and isotype-payload controls (Figure 4, and data not shown). Full datasets across additional PDXO models are presented in the accompanying AACR 2025 poster. These results highlight the utility of PDXO models derived from Champions’ CTG-3D platform for evaluating next-generation ADCs and discriminating between payload-driven cytotoxicity, nonspecific conjugate effects, and HER2-targeted ADC activity. To further quantify differential treatment responses, normalized Area Under the Curve (∆AUC) values were calculated to capture response differences between isotype controls and ADCs across the full concentration range. HER2-positive breast cancer cell lines and PDXO models exhibited higher ∆AUC values (approximately 30–50%, data not shown), indicating stronger ADC activity relative to controls. In contrast, HER2-negative models showed substantially lower ∆AUC values (~20%), consistent with reduced ADC activity in the absence of target expression (Figure 5). Figure 5. Normalized ∆AUC. The response difference between isotypes and ADCs is calculated as: [AUC of Isotype] – [AUC of ADC] / [AUC of Isotype ] x 100% Collectively, these findings demonstrate that breast cancer PDXO models generated from Champions’ CTG-3D platform reproduce expected response patterns to FDA-approved HER2-targeted ADCs with known pharmacological profiles, supporting their utility as a predictive ex vivo platform for ADC development. Key Takeaways Champions’ CTG-3D platform is a biologically relevant ex vivo 3D platform that can support critical preclinical decisions, including payload comparison and mechanism-based screening, enabling ADC candidates to be screened and prioritized before more expensive in vivo evaluation and clinical development is undertaken. For drug developers, this translates into greater confidence in candidate selection, improved translational relevance of preclinical data, and reduced risk of late-stage attrition of potentially viable candidates. By enabling better-informed decisions earlier in the development process, the CTG-3D platform helps accelerate the advancement of safer and more effective ADC-based therapies. To explore the full datasets in more detail, download the full poster presented at AACR 2025 here. References Joubert, Nicolas, Alain Beck, Charles Dumontet, and Caroline Denevault-Sabourin. 2020. “Antibody-Drug Conjugates: The Last Decade.” Pharmaceuticals (Basel, Switzerland) 13 (9). https://doi.org/10.3390/ph13090245.
Evaluating a Novel GCN2 Inhibitor for Acute Myeloid Leukemia (AML) Treatment Using Patient-Derived Models
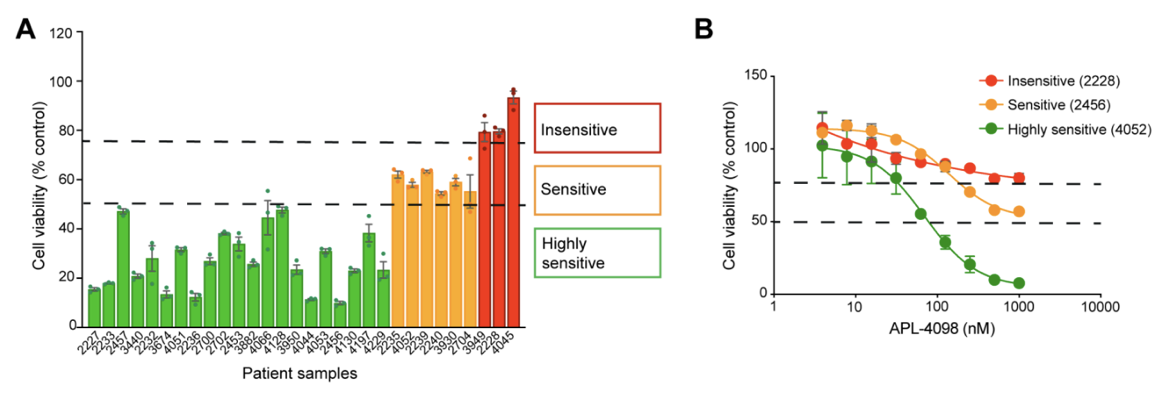
The AML Therapeutic Landscape Relapse remains one of the greatest challenges in treating Acute Myeloid Leukemia (AML). While targeted therapies have improved outcomes for some patients, high relapse rates and limited durable treatment options mean that many patients still face poor prognoses. One pathway gaining attention is the Integrated Stress Response (ISR), a highly conserved protective signaling network that helps cells adapt to nutrient deprivation, oxidative stress, and other metabolic stressors. Many tumor types, including AML, depend heavily on this stress-response system to survive and proliferate. Within this pathway, the kinase GCN2 (General Control Nonderepressible 2) serves as a key sensor of amino acid deprivation and metabolic stress, helping cancer cells survive under unfavorable conditions where lack of exogenous nutrients may disturb intracellular homeostatic balance. Disrupting this survival mechanism has emerged as a promising strategy to selectively target cancer cells and improve treatment outcomes. A recent study explored a novel and selective inhibitor of the stress-response kinase GCN2, APL-4098, as a potential therapeutic approach for AML1. Ex Vivo Evaluation of APL-4098 in Patient-Derived AML Cells Román-Trufero et al. first characterized the anti-leukemic activity of APL-4098 in ex vivo primary patient AML samples using Champions Oncology’s AML VitroScreen platform. A cohort of 30 patient-derived samples was treated with increasing concentrations of the compound and cell viability was measured using a luminescence-based viability readout to determine how effectively the drug inhibited leukemia cell growth. From these results, dose–response curves and IC50 values were generated to quantify drug potency across the cohort. The results revealed strong inhibitory activity across the patient cohort with 70% of samples (21/30) showing a ≥50% reduction in cell viability, indicating high sensitivity to the GCN2 inhibitor. An additional 20% of samples demonstrated some sensitivity, with a 25–50% reduction in viability. Finally, 10% of the samples showed less than 25% decrease in viability and were categorized as insensitive (Figure 1). Figure 1. APL-4098 cytotoxic activity ex vivo. (A) Effect of 500 nM APL-4098 on the viability of patient-derived AML samples after 72h treatment. Values for each specimen are expressed relative to each sample’s matched vehicle-treated control (normalized to 100%) and presented as mean ± SEM, n=3. (B) Selected APL-4098 dose-response curves of highly sensitive (green), sensitive (orange), and insensitive (red) patient-derived samples. Notably, responses were observed across diverse genomic backgrounds, with no clear correlation between common AML mutations and sensitivity. Cells from pre-treated or relapsed patients also responded, suggesting that GCN2 inhibition may be effective even in difficult-to-treat AML. This ex vivo screening step provided critical insight into which patient samples were most sensitive, laying the groundwork for subsequent in vivo evaluation in patient-derived xenograft (PDX) models. In Vivo Evaluation of APL-4098 in AML Patient-Derived Xenografts Following ex vivo screening, the anti-leukemic activity of APL-4098 was further assessed in Champions’ patient-derived xenograft (PDX) AML models. Human AML cells from a patient sample that showed ex vivo sensitivity to APL-4098 were implanted into immunodeficient NOG mice via tail vein injection, allowing leukemia to engraft in the bone marrow. Engraftment of human AML cells in the bone marrow was monitored in surrogate animals using flow cytometry, and once engraftment reached ~20% human CD45+ cells, mice were randomized into four treatment groups: Vehicle control (DMSO) APL-4098 alone (15 mg/kg once daily) Venetoclax alone (100 mg/kg once daily), as a standard-of-care comparator APL-4098 + venetoclax combination Treatments were administered daily for 19 days, and leukemia burden was evaluated by analyzing multiple AML subpopulations, including blasts, progenitors, and leukemia stem cells (LSCs), using multicolor flow cytometry (Figure 2). This allowed researchers to evaluate both single-agent activity and potential synergy with venetoclax in a clinically relevant AML model. Figure 2. Effect of APL-4098, Venetoclax and the combination of both in vivo in an AML PDX model. Bone marrow from AML-engrafted animals was assessed for (A) the percentage of viable human CD45+. (B) the number of viable blasts (CD34+/CD33+/-). (C) the number of viable AML progenitors (CD34+/CD38+) (D) the number of viable LCSs (CD34+/CD38-). In all graphs, data shown as mean ± SEM, statistical significance determined by one-way ANOVA. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. DMSO, n=6; APL-4098, n=7; Venetoclax, n=8, APL-4098 + Venetoclax, n=7. The results showed APL-4098 as a monotherapy had little to no effect on bulk leukemic burden, but preferentially targeted and reduced the LSC-enriched compartment, a niche generally considered primarily responsible for relapse initiation after bulk eradication of leukemic blasts2 Importantly, the combination of APL-4098 and Venetoclax, the standard of care BCL-2 inhibitor in AML patients, produced a pronounced synergistic response, achieving a 97% to 99% reduction across all leukemia subpopulations analysed. Mechanistic studies, including RNA sequencing and metabolic analyses, suggested that APL-4098 exerts its anti-leukemic activity by suppressing mitochondrial respiration, triggering the mitochondrial unfolded protein response, and inducing metabolic stress in AML cells. Together, the researchers demonstrated APL-4098 effectively induces cytotoxicity both ex vivo and in vivo, with preferential effect against the LSC subpopulation, a key driver of patient relapse. While APL-4098 shows potential as a monotherapy agent in the preclinical setting, the potent synergistic interaction between APL-4098 and Venetoclax highlights a potential combination therapy for AML that merits further non-clinical and clinical exploration. This study demonstrates the utility of Champions Oncology’s patient-derived platforms for preclinical and translational AML research, providing a framework to evaluate drug efficacy and explore new therapeutic strategies. To learn more about this study, download the full publication here Explore the Champions Oncology website to discover more about our hematological cancer models and testing capabilities References Román-Trufero M, Whitlock G, Seydoux C, et al. A novel potent and selective GCN2 inhibitor, APL-4098, has anti-leukemic activity through dysregulation of mitochondrial function. Clin Cancer Res. 2026. Hansen Q, Bachas C, Smit L, Cloos J. Characteristics of leukemic stem cells in acute leukemia and potential targeted therapies for their specific eradication. Cancer Drug Resist. 2022.
Not All Tumors Look Alike: Using Surfaceome Diversity to Enrich Responders
The case for measuring the surface directly Biologics and antibody drug conjugates operate at the cell's surface, yet many programs still guide patient selection using RNA expression or whole cell protein abundance. That practice can misclassify candidates because transcripts and total protein often do not correlate, due to post-transcriptional and post-translational mechanisms. Moreover, cellular localization and receptor density can’t be quantified by using whole cell bulk proteomics, therefore not guaranteeing that a receptor is exposed on the exterior membrane above drug relevant thresholds. In 2024, investigators profiled the surfaceome of 100 genetically diverse, primary human AML specimens and resolved antigen patterns on primitive and stem like cells with limited expression in essential normal tissues. The work demonstrated that surface level heterogeneity is real, clinically meaningful, and different from what one would infer from bulk RNA or whole cell proteomics alone. Cell+1 Calls to better map the surfaceome are growing. A 2024 Cancer Discovery commentary noted that only a few dozen cell surface targets currently anchor FDA or EMA approved therapies, despite the surface being the most accessible compartment for antibodies, CAR T cells, and radiopharmaceuticals. The authors argued for intensified efforts to chart the universe of surface proteins across cancers, which would accelerate target discovery and improve translational relevance for modalities that require true surface accessibility. AACR Journals+1 Solving the Surfaceome Problem Why a surface resolved view changes development plans Modern reviews describe how advances in mass spectrometry, fractionation, and enrichment are making it feasible to survey the cancer surfaceome at scale. These approaches improve the identification of drug accessible proteins that whole cell proteomics can miss, because abundant intracellular proteins dominate unfractionated measurements and mask low abundance, surface localized receptors. The reviews also outline practical ways to mitigate technical challenges such as low copy number, hydrophobic domains, and contamination by intracellular compartments, for example through careful membrane preparation, parallel intracellular and whole cell fractions, and stringent enrichment and quality control thresholds. The program level implication is straightforward. When a therapy acts at the surface, the biomarker strategy should resolve surface localization directly rather than infer it from RNA or whole cell protein alone. PubMed+1 A second implication concerns internalization and trafficking. For ADCs and some bispecifics, internalization kinetics and routing to lysosomes affect payload delivery and potency. Surface resolved workflows can be coupled to orthogonal assays, for example flow cytometry with ligand induced internalization or live cell imaging, to determine whether a receptor is not only present at the surface but also behaves in a way that supports the intended mechanism. This behavioral dimension rarely appears in transcript based selection but influences both efficacy and safety. From heterogeneity to practical enrichment Turning surface heterogeneity into clinical signal requires a sequence of disciplined steps. First, measure what matters. Use fractionated surface proteomics or validated surface specific immunohistochemistry and flow assays that distinguish the exterior membrane from total abundance. Second, define actionable thresholds that tie expression to benefit and that can be reproduced across sites. Educational content from the ASCO Educational Book emphasizes that quantitative cutoffs and standardized assays are central to patient selection for antibody drug conjugates, because target accessibility and abundance determine benefit, and because inconsistent thresholds erode the interpretability of early phase studies. Third, map prevalence by histology and by clinically relevant subgroups, since an attractive target with low prevalence may not support enrollment or may require a targeted site strategy. When these elements are present, enrichment reflects how the drug actually works and not a proxy signal. ASCO Publications+1 Surface level diversity can also sharpen indication strategy. If a receptor is highly enriched at the surface in a subset of colorectal cancer but not in pancreatic cancer, a program can prioritize the former for first in human evaluation, even if RNA levels are similar in both. This is particularly relevant where surface and whole cell abundances diverge. The AML study noted above showed precisely this phenomenon, where antigen exposure on stem like compartments could be quantified and connected to therapeutic concepts that require surface binding. Cell Selectivity and safety as design constraints Surface heterogeneity intersects with safety because many candidate antigens have some expression in normal tissues. A 2024 review in Trends in Pharmacological Sciences surveyed strategies that increase antibody selectivity in oncology, including superselectivity through avidity and multivalency, conditional or pH sensitive binding, dual targeting that requires co expression to achieve high affinity, and engineering designs that leverage tissue context. These concepts translate directly into earlier program decisions. Targets that can be paired with selective engineering approaches should score higher than those that would require unrealistic discrimination from a conventional binder. A transparent scoring framework, for example one that penalizes normal tissue expression in essential organs and rewards tumor specific co expression patterns, makes those choices auditable during governance reviews. Cell+1 The safety argument is not theoretical. ADCs can cause off tumor toxicities when a payload is delivered to normal tissues that express the target at modest levels. A biomarker plan that quantifies true surface exposure in disease and screens for surface exposure in a curated panel of normal tissues improves the chance of a workable therapeutic window. Reviews that focus on ADC biomarkers, together with trial design guidance, point in the same direction, namely that selection should be quantitative and assayable, not an exploratory cutpoint chosen after the fact. ASCO Publications Concrete examples of surfaceome driven discovery Evidence that surface resolved discovery translates into programs is accumulating. In late 2024, a Cancer Cell study used an integrative proteogenomic surfaceome approach to credential DLK1 as an immunotherapeutic target in neuroblastoma. The authors combined mass spectrometry based surface profiling with genomic and transcriptomic context to demonstrate surface exposure, tumor specificity, and functional relevance, then moved to validation experiments that supported drug development. This work illustrates how surface level datasets, anchored in clinical material, can identify and qualify targets that a transcript only screen might underrate. PMC+3Cell+3PubMed+3 Surfaceome maps are also becoming more accessible, which will help teams generalize findings across institutions. The AML dataset is public in GEO, enabling independent inspection of QC criteria, antigen lists, and statistical methods. Commentary from Cancer Discovery underscores the systemic opportunity, arguing that better cartography of the surfaceome across cancers is likely to grow the small set of currently actionable surface targets. Together, public data and field level commentary support a move from opportunistic target picking toward systematic, population informed discovery. NCBI+1 TALK Decoding the Cell Surface to Accelerate Discovery Program design, trial execution, and measurement Programs that embrace surface resolved selection can operationalize three habits that improve downstream signal. First, connect discovery assays to clinical screening early. If discovery uses a fractionated surface proteomics threshold, define the clinical assay that will mirror that readout, for example an IHC score or a validated flow protocol, and harmonize cutoffs before first patient in. Second, pre specify enrichment rules in the protocol. Enrolling only the top percentiles of surface expressors may seem restrictive, but it increases the chance of a pharmacodynamic signal that validates the mechanism and informs dose expansion. Third, measure what happens when selection criteria are varied in sensitivity analyses, and share these details in publications and regulatory interactions. This transparency builds cumulative knowledge that benefits the field and informs the next iteration of thresholds. There is also value in linking surface metrics to pharmacology. If a surface antigen is abundant but shows slow internalization, a payload with a bystander effect may be preferable to a payload that requires rapid lysosomal routing. If surface expression is heterogeneous at the lesion level, a radionuclide therapy that can exploit crossfire may offer advantages over a conventional ADC. These are not general rules, they are examples of how a surface resolved view can shape the choice of modality and payload in a way that reflects the physical constraints of the target. Where Champions Oncology contributes Champions Oncology generates surface resolved datasets from low passage, clinically relevant tumor models and integrates them with deep multi omic profiles and in vivo pharmacology. This enables teams to quantify true surface positivity, to set and test actionable thresholds, and to understand prevalence by indication before committing to costly trials. The same datasets support orthogonal validation and method standardization so that discovery assays translate into clinical screening. The approach is designed to be non promotional and data first, the objective is to clarify risk and to help programs make better decisions earlier.
Beyond Single Omic Biomarkers: How Proteogenomic ML Reveals Therapy Vulnerabilities
Why a functional view changes predictions Genomics and transcriptomics remain foundational for precision oncology, but they do not fully represent the functional state that determines how tumors respond to therapy. Proteins and phosphoproteins capture activity at the level where drugs actually engage, for example receptor density and localization, complex assembly, and pathway signaling. That distinction is not academic. In a 2024 pan-cancer analysis from the Clinical Proteomic Tumor Analysis Consortium (CPTAC), investigators integrated proteogenomic data from 1,043 patients across 10 tumor types, surveyed 2,863 druggable proteins, and quantified biological factors that weaken mRNA to protein correlation, making the case for models that learn directly from protein and phosphoprotein context rather than inferring from transcripts alone. Cell From data to models that travel The practical bottleneck has been access to harmonized, well-annotated cohorts that support training, testing, and independent validation. In August 2023, the National Cancer Institute announced a standardized pan-cancer proteogenomic dataset that aligns genomics, proteomics, imaging, and clinical data for more than 1,000 tumors across 10 cancer types, explicitly to enable reproducible discovery and model benchmarking. The Proteomic Data Commons (PDC) now serves these resources in a way that supports programmatic access and cross-study comparisons, a requirement if machine-learning outputs are going to generalize beyond a single study. National Cancer Institute+1 Solving the Surfaceome Problem What the evidence shows when proteins are included Two Cell papers from 2024 illustrate why adding protein-level information changes conclusions. An immune-landscape analysis derived distinct immune subtypes by integrating genomic, epigenomic, transcriptomic, and proteomic features and connected oncogenic drivers to downstream protein states that influence immune surveillance and evasion. A companion pan-cancer study expanded the landscape of therapeutic opportunities by evaluating thousands of druggable proteins across tissues and documenting where mRNA is a poor proxy for protein, especially in pathways relevant to therapy response. Together, they show that multi-omic modeling, including protein and phosphoprotein features, improves biological interpretability and exposes actionable biology that single-omic approaches overlook. Cell+1 A broader signal from the field The trend is not confined to CPTAC. The Pan-Cancer Proteome Atlas (TPCPA), published in Cancer Cell in 2025, quantified 9,670 proteins across 999 primary tumors representing 22 cancer types using DIA-MS. The atlas offers a tissue-based substrate for target nomination, biomarker discovery, and external validation, and has been highlighted in the trade press for its global availability and immediate relevance to oncology research. Such atlases are valuable because they capture proteomic variability directly in clinical material, not only in cell lines, providing realistic distributions for features that ML models attempt to learn. Cell+2PubMed+2 Why proteogenomic ML improves prediction Integrating proteins and phosphoproteins adds information that is both mechanistic and measurable. First, pathway activity is reflected in phosphorylation states, which function as on–off or rheostat-like controls for signaling. Second, receptor exposure and complex formation at the protein level determine whether a therapy can bind or disrupt a process. Third, protein degradation and post-translational regulation often decouple mRNA abundance from target availability, which explains why transcript-only biomarkers can fail at the bedside. When these features are engineered into models, performance gains are not just numeric; they tend to be more interpretable, mapping to drug-actionable pathways and receptors that clinicians recognize. The 2024 CPTAC studies provide concrete examples. Immune subtypes defined by proteogenomic features correlate with differences in antigen presentation, cytokine signaling, and interferon responses, features with obvious translational implications. The survey of druggable proteins shows wide variation in abundance and localization across tumors and details the contexts where transcript and protein diverge, arguing for protein-aware rules when nominating targets or stratifying patients. Cell+1 What good practice looks like in model building There is a growing consensus on practical guardrails. Independent validation across cohorts is essential to avoid overfitting, and the infrastructure now exists to support that step through the PDC and related CPTAC resources. Feature construction should prioritize pathway-level signals that aggregate individual phospho-sites into kinase or pathway activity because these are more stable across cohorts and easier to interpret for clinical decision making. Finally, clinically annotated samples, including treatment history and outcomes, are indispensable if models are expected to inform responder enrichment and mechanism-of-resistance hypotheses rather than only classify molecular subtypes. National Cancer Institute Translational payoffs, with appropriate caution When executed with these guardrails, proteogenomic ML offers tangible benefits. Programs can generate earlier responder and non-responder hypotheses and test them prospectively in preclinical systems before committing costly clinical designs. Resistance pathways inferred from phospho-proteomic features can motivate combination strategies, for example pairing an antibody-drug conjugate with a kinase inhibitor when signaling indicates a plausible escape route. Educational content from ASCO has emphasized the centrality of quantitative thresholds and validated assays for patient selection, particularly for ADCs where surface accessibility and abundance determine benefit. The lesson is consistent across modalities. Predictive features must be connected to assays that can be deployed consistently in trials, and thresholds should be defined in a way that anticipates real-world variability. PubMed Where limitations still matter Several limitations deserve explicit mention. Proteomic and phospho-proteomic data remain technically variable across platforms and laboratories. Although CPTAC and PDC mitigate this through standardization, modelers should evaluate batch effects and apply normalization strategies suited to proteomic data. Coverage of kinase–substrate relationships and post-translational networks is incomplete, which constrains inference. Tumor heterogeneity adds another layer, particularly when bulk tissue averages mask subclonal or microenvironmental signals. These caveats do not negate the value of proteogenomic ML, but they do argue for conservative claims, orthogonal validation, and a bias toward features that can be measured reproducibly in clinical settings. TALK Decoding the Cell Surface to Accelerate Discovery Implications for trial design and portfolio focus The immediate implication is a more disciplined approach to enrichment. If protein-level features identify a subgroup with plausible sensitivity, early designs can incorporate eligibility criteria and stratification based on validated assays rather than exploratory cutpoints. Conversely, if pathway-level features suggest multiple escape routes, it may be more efficient to prioritize combinations earlier instead of iterating single-agent studies. At a portfolio level, proteogenomic evidence can help prioritize programs with a mechanistic rationale supported by functional data, not only by mutation prevalence or gene expression. How Champions Oncology contributes Champions Oncology builds models on tumor-derived systems that preserve patient biology and heterogeneity. Our datasets link genomics and transcriptomics with proteomics, phospho-proteomics, and cell surface proteomics, and they are annotated with pharmacologic phenotypes. This combination supports models that tie features to functional biology and drug accessibility, making it possible to move from correlation to causal relation and from causal relation to druggable targets.

Feature selection in RNA-seq and proteomics with MADVAR in R
High dimensional omics datasets often include many features that contribute little to downstream analysis. This can blur structure in unsupervised tasks, slow computation, and complicate model training. The MADVAR study introduces two simple, data driven procedures that set feature selection thresholds from the distribution of the data itself, rather than relying on fixed heuristics. The first procedure, madvar, computes a variance cutoff using the median plus a multiple of the median absolute deviation. The second, intersect Distributions, fits a two component Gaussian mixture to the variance or another continuous score, and uses the intersection point between components as the cutoff. Both methods are implemented in an R package and were evaluated across public datasets that include TCGA gene expression, GTEx proteomics, and CPTAC phosphoproteomics. The paper reports improvements in unsupervised clustering quality and competitive supervised performance with fewer features, while keeping runtime and memory use modest. What the Paper Tested The benchmarking examined unsupervised structure and supervised classification. For unsupervised analysis, the authors applied filtering and then assessed cluster quality with connectivity, the Dunn index, and the Biological Homogeneity Index. Across datasets, the variance based approaches produced tighter or more homogeneous clusters on these metrics. For supervised analysis, they trained random forest models with repeated runs. Both approaches produced low out of bag error rates. Retaining more features sometimes improved accuracy, but MADVAR often matched the mixture based approach while selecting fewer features. The paper also documents practical defaults, such as Ward.D for hierarchical clustering with Euclidean distance, and explains how to pass either a raw matrix or a precomputed variance vector into the functions. Source code and documentation are available on GitHub. When These Methods are a Good Fit These procedures are particularly well suited for rapid, large-scale preprocessing, when analyses require a quick, efficient, and transparent approach to feature selection prior to dimensionality reduction, clustering, or model fitting. They also integrate naturally into interpretability-oriented pipelines, since thresholds based on medians or mixture-model intersections are simple to explain and justify to collaborators. Because the logic operates on continuous feature scores, the same framework can be applied seamlessly to any quantitative data type that can be summarized by variability Things to keep in mind. Variance is a proxy for informativeness, not a guarantee. Low variance does not always mean a feature is uninformative. Some biomarkers remain stable yet become predictive in combination with others. If domain knowledge indicates a feature should be preserved, the package allows must keep lists. The mixture based method assumes the variance distribution resembles a two-component mixture. If the fit is poor, the intersection may not be meaningful, so density plots are worth inspecting before adopting the cutoff. Downstream metric choice also matters. Gains in the Biological Homogeneity Index or Dunn index describe cluster characteristics, which may not translate to improvements on other endpoints such as survival modeling or dose response prediction. Finally, supervised performance can depend on class imbalance and sample size. If your data are skewed, tune the learner and validate with a scheme that reflects your use case. Read the Full Paper How to Apply, a Straightforward Workflow A practical workflow starts by exploring the variance distribution. Plot the density (using the madvar flag `plot_density = TRUE`), confirm whether there is a near zero peak, and decide whether a MAD based threshold or a two-component mixture are appropriate. Set a conservative first pass using the default MAD multiplier (`mads = 2`) and adjust if another stringency level is preferred, or, if you prefer the mixture approach, verify the intersection visually before you commit to the cutoff. Preserve domain critical features by whitelisting known markers or controls that should not be dropped. Re run the planned clustering or model fitting, compare structure and error rates before and after filtering, and record any change in feature counts and compute time so the impact is transparent to collaborators. Reproducibility and availability The R implementation and documentation are available on GitHub, as referenced in the paper. The evaluations draw on TCGA gene expression, GTEx proteomics, and CPTAC phosphoproteomics, with figures that show density plots, clustering metrics, and classification results. The article appears as an Application Note in Bioinformatics Advances and is available for open access. Summary MADVAR provides two transparent, variance-based rules for feature selection, enabling the removal of near invariant features from large omics matrices. In the reported benchmarks, these procedures improved or maintained clustering quality and supervised accuracy while substantially reducing feature counts and computational load. The approach is easy to inspect, easy to explain, and simple to integrate into existing R workflows. As with any filter, final value depends on the analysis goal, so it is worth validating its effects on the endpoints that matter for your study. Silberberg G. “MADVAR, a lightweight, data driven tool for automated feature selection in omics data.” Bioinformatics Advances 2025, vbaf211. doi, 10.1093/bioadv/vbaf211. Explore our Data Ecosystem

Choosing the Right CDX Models: Speed, Scientific Value, and Translational Relevance
In oncology drug development, cell line-derived xenograft (CDX) models remain one of the cornerstone in vivo tools for evaluating new therapies in the preclinical ecosystem. They offer a balance of biological relevance, reproducibility, and speed that makes them ideal for early phase hypothesis testing. Nevertheless, while CDX models are indispensable for initial target screening and validation, they also have well-understood limitations that can upset downstream progress if not properly accounted for. As drug developers increasingly rely on CDX-based systems to screen and prioritize compounds before moving into clinically relevant, albeit more expensive, patient-derived xenografts (PDX), the need for appropriate model selection has become critical. The right CDX program can deliver early translational clarity and strategic focus, while the wrong one can generate noise that obscures the true efficacy of an otherwise promising therapeutic, potentially derailing its continued development. Balancing Speed and Fidelity CDX studies are often designed for speed. They enable large-scale screening of novel agents, as both mono- and combination therapies, and dosing regimens in a fraction of the time and cost required for PDX studies. This makes them especially valuable at the earliest stages of decision-making, where timelines are incredibly compressed and attrition risk is high. But that same speed exacts a cost. Many CDX models originate from long established cell lines that have been maintained in culture for decades. Over time, these lines lose virtually all the critical genomic and cellular heterogeneity, stromal interactions, and microenvironmental complexity characteristic of a bona fide tumor. Whilst they grow predictably, the biological features they retain may no longer reflect that of tumors seen in clinical patients. However, the key is not to abandon CDX models, but to recognize where they fit in the development pipeline and to acknowledge and mitigate the limitations they have. Used prudently, CDX models are an efficient and scientifically powerful system to rank compounds, explore mechanisms, and refine hypotheses before moving into PDX for deeper translational validation. Defining a High Value CDX Model The quality of a CDX model is defined by its biological fidelity and characterization depth. Models derived from contemporary, clinically annotated cell lines are more likely to capture the genomic and phenotypic diversity relevant to modern oncology therapeutics. When models reflect the intrinsic complexity of current patient populations, such as the plethora of KRAS G12 mutations or the legion mechanisms by which EGFR becomes hyperactivated, they offer drug developers meaningful results and mechanism-linked insights that can inform clinical strategy. Biological and “omic” characterization matters as much, if not more, than cellular/tissue origin in CDX models, particularly as clinical oncology continues to diverge from tissue-based therapy to therapeutic roadmaps grounded in the molecular features of each patient tumor. A high value CDX model is supported by multi-omic profiling that includes genomic, transcriptomic, and proteomic annotation. This data allows researchers to interpret observed drug effects through the lens of pathway activation, resistance mechanisms, and biomarker expression. In contrast, models lacking such characterization risk generating results that are descriptive rather than explanatory. Drive Proof of Concept and Target Validation with Champions’ CDX Models Mechanistic Clarity Through Multi-Omic Integration The rise of precision oncology has shown that pharmacology and data science are inextricably linked. Researchers now expect and rely on preclinical models to yield mechanistic understanding and insight, not just tumor regression rates. Integrating omic datasets into CDX studies transforms them from mere screening tools into translational resources capable of generating biological comprehension and preclinical direction. For instance, RNA sequencing of treated and untreated xenografts can reveal transcriptional signatures underpinning positive response outcomes, potentially allowing clinical partitioning of patients likely to receive the most benefit from a therapy. As another example, phosphoproteomic profiling can identify compensatory signaling cascades mediating adaptive drug resistance, permitting de novo combination therapies to be trialed to preempt such resistance before it evolves. This approach enables drug developers to anticipate how tumors might evade inhibition, long before clinical exposure. Moreover, omic integration provides a framework for cross platform alignment. Data from CDX models can be mapped against PDX datasets, public repositories, or patient trial cohorts, accelerating the feedback loop between preclinical findings and clinical validation. Modeling Resistance and Tumor Evolution One of the most powerful applications of CDX technologies is in modeling acquired resistance. By exposing tumor-bearing mice to sustained drug pressure, scientists can select for resistant clones that mimic clinical relapse. Comparative molecular profiling between parental and resistant CDX lines may illuminate the pathways that drive therapeutic escape, whether through secondary genomic changes, activation of bypass signaling cascades, or metabolic rewiring. This approach supports the rational design of next-generation inhibitors or combination strategies aimed at delaying or overcoming resistance. It also informs biomarker development by revealing the early molecular changes that forecast reduced drug sensitivity, enabling the design of clinical trials with built-in resistance monitoring. Bridging the Gap to PDX Despite their wide-ranging utility and flexibility as experimental tools, CDX models are at best a starting technology in the developmental pipeline. Translationally-minded organizations deploy CDX models as a filter for promising candidates in an experimental continuum that leads naturally into PDX. PDX models, established directly from patient tumors, preserve the architecture, stromal components, and molecular heterogeneity of the original cancer. They capture biological features that CDXs inherently lack, including contextualized immune responses in humanized systems, evolution and expansion of tumor sub-clones, and the influence of the microenvironment on cancer progression. For these reasons, PDX validation remains an important next step, if not a critical one, once a compound demonstrates clear activity in CDX. The most efficient development pipelines are those where CDX and PDX models are in biological alignment, where both originate from well characterized sources and overlap genomically and/or phenotypically. In this context, compound evaluation flows smoothly from screening to translational evaluation. A consistent molecular linkage between model systems strengthens the predictive bridge and ensures that early results translate more faithfully into clinical outcomes. Speed Without Sacrificing Relevance The enduring appeal of CDX model systems lies in the speed with which large quantities of data can be generated to reinforce or oppose development of individual drug compounds, or indeed entire drug programs. Studies can be initiated quickly, with timelines to results measured in weeks rather than months. Moreover, CDX models can support the simultaneous exploration of multiple therapeutic hypotheses. Where delays mean patients remain beset with therapeutic inadequacies and can cost developers millions in lost opportunity, this speed is a major competitive advantage. But whilst speed is a necessary component of drug development, it is insufficient for success. The most effective drug pipelines are designed with translational intent incorporated into CDX programs from the outset. CDX model selection is based on mechanistic alignment derived from deep omics comprehensive characterization, and the clinical transition to PDX model systems is a crucial element of the sequence rather than an unintegrated effort. This approach ensures CDX models are deployed as value-enhancing study tools,leveraging the efficiency of CDXs to inform smarter, faster progression into the PDX models best approximating patient clinical features and responses. Designing a Modern CDX Strategy For biotechnology and pharmaceutical teams, a modern CDX strategy balances three principles:speed, depth, and connectivity. Speed means using CDX models to quickly triage candidate molecules, confirm on-target effects, and eliminate ineffective compounds prior to larger resource investment. Depth refers to multi-layered omics characterization to uncover mechanistic drivers of response or resistance. And finally, connectivity means designing CDX studies with the downstream transition to PDX models in mind, ensuring molecular alignment and continuity between the different systems. When these principles are applied judiciously, CDX models become a strategic asset that accelerates development timelines without compromising scientific integrity. The Future of Translational Modeling As oncology continues to evolve toward precision medicine, the most impactful preclinical programs will be those that connect fast data generation with deep omics characterization, using rapid CDX screening to guide more complex, patient-relevant studies. Emerging approaches such as multi-omic analytics, AI-driven model selection, and ex vivo/organoid validation are expanding how CDX data can inform clinical decision-making. Taken in concert, these all suggest a future where the value of a model is defined not only by how fast it can be employed to produce data, but also by how that same data can be used to map the necessary downstream steps to ensure successful drug development and patient application. Choosing the Right Partner In an increasingly competitive preclinical landscape, the distinction between a vendor and a scientific partner has never been clearer. The most valuable CDX programs combine biological relevance, data transparency, and translational foresight. When evaluating potential collaborators, sponsors should ask not only what models are available but how those models were developed, characterized, and validated. The answers will reveal whether a platform can deliver more than results, whether it can deliver understanding. Cell Line Select Tool A smarter way to explore Champions’ cell line models.