
Trends in Oncology
A Needle in a Haystack: Finding Rare AML Populations by Flow Cytometry
Hematologic malignancies include a wide array of lymphomas and leukemias that affect different immune cell subsets. Acute myelogenous leukemia (AML) is one of the most commonly occurring leukemias in adults and children. AML is a highly heterogeneous disease that can be caused by spontaneous gene mutations or chromosomal translocations, which result in the proliferation of dysfunctional myeloid cells. Cytogenetic and morphologic analyses have been the gold standard methods used in AML diagnosis. Still, flow cytometry-based protocols are becoming more widely used and validated as complementary diagnostic methods that can be coupled with these analyses to better guide treatment plans. Flow cytometry has also become an essential tool to understand AML progression and develop and evaluate novel therapeutics. Consider these aspects of flow cytometry-based analysis of AML for exploratory or preclinical research: Phenotype matters: Immunologists know a lot about what a normally developing myeloid cell should look like in terms of its immunophenotype. Changes in expression of different lineage markers correlate strongly with AML progression and are characterized by the presence of blasts (leukemic cells) in the bone marrow. Flow cytometry-based immunophenotyping offers researchers a rapid and sensitive method for detecting blasts at the onset of disease as well as monitoring changes in this population throughout an experimental therapy. Rare cells from precious samples: A major consideration of following AML progression is the ability to detect relatively rare cells in bone marrow aspirates. This type of sample may be relatively small and is typically used fresh for morphologic evaluation, but even a small volume of remaining aspirate can be used for flow cytometry-based methods that are sensitive and robust enough to detect blasts. Paired samples: Bone marrow cells can be analyzed along with peripheral blood using advanced flow cytometry methods that monitor the persistence of blasts and other leukemic subsets over time in these compartments. This type of analysis offers critical insight into the development of new therapeutics whether they are being evaluated in humanized mouse models or clinical trial participants. Flow cytometry continues to advance immuno-oncology research, especially for diseases involving the detection of rare cell populations. Consider working with preclinical and clinical flow cytometry experts to develop protocols for future immuno-oncology studies.
Enhancing CAR T Cell Therapy: Optimizing Preparation for Superior Results
Chimeric antigen receptor-mediated T cell (CAR T cell) therapies have revolutionized the treatment of hematologic malignancies and solid tumors. This therapy uses T cells, typically harvested from patients, that are engineered to express chimeric antigen receptors (CAR) specific to tumor cell antigens. CD19-targeting CAR T cell therapy was the first immunotherapy shown to effectively treat acute lymphoblastic leukemia[1], but a subset of patients relapse due to loss or poor engraftment of CAR T cells. Here we highlight advances in CAR T cell therapy to improve the quality of the immunotherapy product ex vivo for more effective responses in vivo. Culture Conditions T cells from peripheral blood mononuclear cells (PBMCs) are the primary cellular product used for CAR T cell therapy, but several steps must be carried out ex vivo to ensure that enough cells are made for therapeutic efficacy. The best treatment outcomes have been linked to high levels of CAR T cell engraftment and persistence upon transfer into a patient[2]. Ex vivo culture methods have been optimized to expand T cells, and cultures using less differentiated T cells, like stem cell memory T cells or naïve-like T cells, have been linked to better persistence upon transfer[3]. Recent studies are characterizing phenotypic features of T cells that preserve “stemness” upon ex vivo culture but still allow for expansion and expression of CAR T receptors. Reduced culture time has been one of the most effective methods for improving engraftment and antitumor responses but is limited by the number of cells yielded by this minimal manipulation process[4]. Biodistribution The successful engraftment and persistence of CAR T cells depend greatly on where cells migrate upon transfer into patients. Preserving stem cell-like features correlates with better engraftment, especially if donor-derived cells are unavailable and autologous cell sources must be used[5]. In vivo imaging studies have been very helpful in understanding CAR T cell dynamics and anti-tumor responses. Studies in mouse models have indicated that CAR T cells can get trapped in tissues, including the lungs, which can limit access to tumor targets[6]. Analysis of CAR T cells with tumor cells also revealed extensive functional heterogeneity, including CAR-T cells that can interact with tumor cells but not exert cytotoxic effector functions. A recent first-in-human study examined the biodistribution of radioisotope-labeled CAR T cells and confirmed that these cells rapidly distribute to tumor tissue but are taken up by the liver and spleen and can persist systemically for up to two weeks[7]. As new in vivo imaging studies are carried out, these insights will inform how CAR T cell products are made and delivered and are likely to improve treatment outcomes. Advances in ex vivo methods for CAR T cell preparation are already improving outcomes, and these methods are broadly applicable to donor-derived or autologous T cell products. In vivo imaging methods are also delineating characteristics of CAR T cells that improve tumor targeting or result in misdirected tissue homing. Future in vivo and ex vivo studies are well-poised to further advance CAR T cell applications.
Unlocking the Potential of CAR-T Therapy for Treatment of Multiple Myeloma
Multiple myeloma (MM) is the second most common hematological cancer[1] and results from a series of mutations that lead to malignant plasma cells in the bone marrow.[2] The survival of MM ranges from 5 to 7 years on average, although patients with high-risk MM, defined as inactive TP53 or 1q21 activation, have reduced survival of 2 years.[3, 4] Due to the complexity of this disease, MM remains incurable, however, the emergence of CAR-T (chimeric antigen receptor T-cell) therapy has provided hope for MM patients. Pathogenesis Under normal conditions, plasma cells secrete polyclonal protein or immunoglobulins comprised of one heavy chain and one light chain.[5] MM is derived from a series of mutations in plasma cells and clonal plasmacytes that instead produce monoclonal proteins (M-proteins) within the bone marrow (BM).[6] This disease typically progresses from monoclonal gammopathy of undetermined significance (MGUS) that later develops into smoldering multiple myeloma (SMM) – both of which are asymptomatic plasma cell disorders with no end-organ damage.[7] MGUS is defined as a lower percentage of M-protein (<3g/dl) while SMM is defined as M-protein concentration exceeding 3g/dl.[8] It is thought that initial mutation events seen in MGUS, and ultimately in MM, that lead to M-protein secretion occur during somatic hypermutation or class-switch recombination events, namely translocations involving IgH locus and IgL locus.[7] The results of these mutations form an abundance of dysfunctional plasma cells within the bone marrow leading to reduced immune system responses and organ damage. Therapeutics Standard of care treatment for newly diagnosed MM includes a cocktail of high-dose chemotherapy agents: bortezomib, lenalidomide, and dexamethasone.[9] These chemotherapeutic agents are effective at damaging tumor cell DNA as well as tumor microenvironment. Following this aggressive combination therapy, patients undergo allogeneic stem cell transplantation (ASCT) to restore blood cell production.[10, 11] Despite the success of these treatments, many MM patients experience graft-versus-host disease (GVHD) and/or relapse,[12] making it essential for new and innovative treatments to emerge. One treatment that has shown promise is chimeric antigen receptor T cell (CAR-T) therapy. CAR-T therapy is a type of immunotherapy that genetically modifies the patient's own T cells to target and kill cancer cells. [12, 13] In MM patients specifically, B cell maturation antigen (BCMA) targeted CAR-T therapy has been highly successful in newly diagnosed and relapsed MM patients. [13] Furthermore, clinical studies have found that CAR-T therapy has a 76% remission rate and conclude better 5-year patient outcomes.[14] Today there are two FDA-approved CART-T therapies for MM, Carvykti and Abecma, with others in development.[15] Despite successes, there are notable challenges with CAR-T therapy including cytokine release syndrome (CRS) and neurotoxicity, which may be managed with appropriate and timely interventions. [13] CAR-T therapy is also costly and requires specialized manufacturing facilities, limiting its availability to patients. Importantly, new treatments outside of CAR-T therapy are also in development with novel targets, namely Selinexor. Selinexor targets Chromosome Region Maintenance 1 (XP01), a transporter of nuclear proteins with a key role in cell cycle regulation and proliferation.[16] XP01 has been implicated in several solid tumors, such as lung cancer, and is overly expressed in MM. Overexpression of XP01 allows tumor suppressor proteins, such as Rb, p53, and p21, to be exported into the cytoplasm rendering them inactive and therefore increasing anti-apoptotic cell signaling.[16, 17] Selinexor covalently binds to the “cargo-binding” groove of XP01 which prevents the displacement of tumor suppressor proteins mentioned above.[17, 18] As a result of its pre-clinical and clinical trial success, the FDA approved Selinexor as a combination therapy with dexamethasone for patients with more than one prior therapy and at least 4 prior dexamethasone therapies.[19, 20] While MM continues to be a challenging cancer to treat, therapies such as CAR-T therapy and Selinexor provide MM patients with new hope for lasting remission. New clinical trials, such as NCT05177536 to test iberdomide maintenance therapy, are underway to explore the efficacy of new therapies and interventions to improve MM outcomes.
Benefits of Immuno-Oncology for Colorectal Cancer
Colorectal cancer (CRC) is a leading cause of cancer deaths globally. Advances in early detection have improved survival rates, but patients diagnosed with metastatic CRC still have stubbornly poor 5-year survival rates. Standard treatments for CRC include surgery, chemotherapy, and radiotherapy, but alternative immuno-oncology therapies are showing promising results in CRC patients. Here we highlight advances in immuno-oncology therapies that are being used to treat CRC patients or are being pursued in preclinical and clinical studies. CRC Subtypes Molecular genetic analysis of CRC tumors is a critical diagnostic tool for classifying and treating this cancer. CRC tumors are typically classified as mismatch repair-deficient/microsatellite instability-high (dMMR—MSI-H) tumors, which have a high overall mutation burden, or mismatch repair-proficient/microsatellite instability-low (pMMR—MSI-L) tumors, which have a lower mutation burden[1]. Defects in MMR are associated with the accumulation of mutations and are caused by defects in mismatch repair proteins, and these defects are typically detected by frame-shift mutations in DNA repeat regions known as microsatellites. Mutations in the BRAF oncogene, particularly the activating V600E mutation, comprise a distinct subset of CRC. BRAF is a component of the mitogen-activated protein kinase (MAPK) pathway that normally functions downstream of the epidermal growth factor receptor to regulate transcription of genes involved in cellular growth and survival, but the BRAF-V600E mutation results in constitutive activation of BRAF and uncontrolled cellular proliferation and tumor growth[2]. Immuno-Oncology Interventions Immuno-oncology approaches that target immune checkpoint blockade have proven effective for several cancers, including CRC. T cells can initiate effective anti-tumor responses under ideal conditions, but the immunosuppressive tumor microenvironment of some cancer types inhibits T cell activation, usually through engagement of immune checkpoint molecules like PD-1 and CTLA4. For more than a decade, the development and use of immune checkpoint inhibitors (ICIs) has transformed the treatment of melanoma[3,4] and non-small cell lung cancer[5,6]. The first FDA-approved treatments include a monoclonal antibody (mAb) that targets CTLA4 (ipilimumab) and two mAbs that target PD-1 (pembrolizumab and nivolumab). These early studies suggested that tumors with high mutation burdens respond well to ICI, which may be due in part to the generation and presentation of tumor neoantigens that are recognized as non-self and can be targeted by cytotoxic T cells[7]. CRC tumors with the dMMR—MSI-H signature have a high mutational burden and typically have a high level of CD4+ and CD8+ tumor-infiltrating lymphocytes (TILs). These cells have shown elevated expression of PD-1, PD-L1, and CTLA4, which suggests that they may respond well to ICIs[8]. Indeed, several recent clinical studies have shown improvements in overall survival and progression-free survival in patients treated with individual or combined ICIs that target PD-1. Of note, interim results from a phase II trial that enrolls patients with MMRd locally advanced rectal cancer to receive neoadjuvant dostarlimab, a PD-1 inhibitor, showed sustained complete response 1 year after the end of treatment in all patients[9]. Other current studies are evaluating next-generation PD-1 inhibitors combined with chemotherapy and/or other biologics such as an anti-VEGF and anti-EGFR mAb[10]. In contrast, patients with pMMR–MSI-L have shown poor responses to PD-1 or CTLA4 blockade alone or in combination, which has led to the development of trials that explore different treatment combinations, including inhibitors of the MAPK pathway or angiogenesis[11]. Promising results have been presented in a phase I/II clinical trial (NCT04017650) in which patients with MSI-L and BRAFV600E were treated with BRAF+EGFR inhibitors, to induce a transient MSI-H phenotype, plus anti-PD-1 antibody[12]. Next-Generation Treatments Several cutting-edge immuno-oncology therapies are being explored for the treatment of CRC. Beyond combinations of individual antibodies, researchers are engineering bispecific antibodies that bind to tumor cells and T cells simultaneously to enhance anti-tumor T cell responses. One such bispecific antibody, CEA-TCB, is being tested in phase I trials alone or in combination with anti-PD-L1 to treat metastatic CRC[13]. Another novel approach includes adoptive cell therapies like chimeric antigen receptor (CAR) T cells, which are T cells collected from the tumor tissue or peripheral blood of a patient and engineered to bind to tumor antigens and potentiate anti-tumor responses. Oncolytic virus and bacteria-based vaccines are also being studied as potential CRC treatments. Besides treatments designed on the tumor's intrinsic genetic background, much effort is being put into deciphering the tumor microenvironment, with a particular focus on its interplay with the gut microbiota to modulate inflammation and immune response, known to be involved in the metastatic onset. These studies are leading to the identification of inflammatory/immune signatures that inform the therapeutic agents to be used to target the pro-oncogenic microenvironment and reactivate the immune system[14]. The advancement of immuno-oncology is already transforming the treatment of CRC and will contribute to better outcomes for all types of CRC in the decades to come.
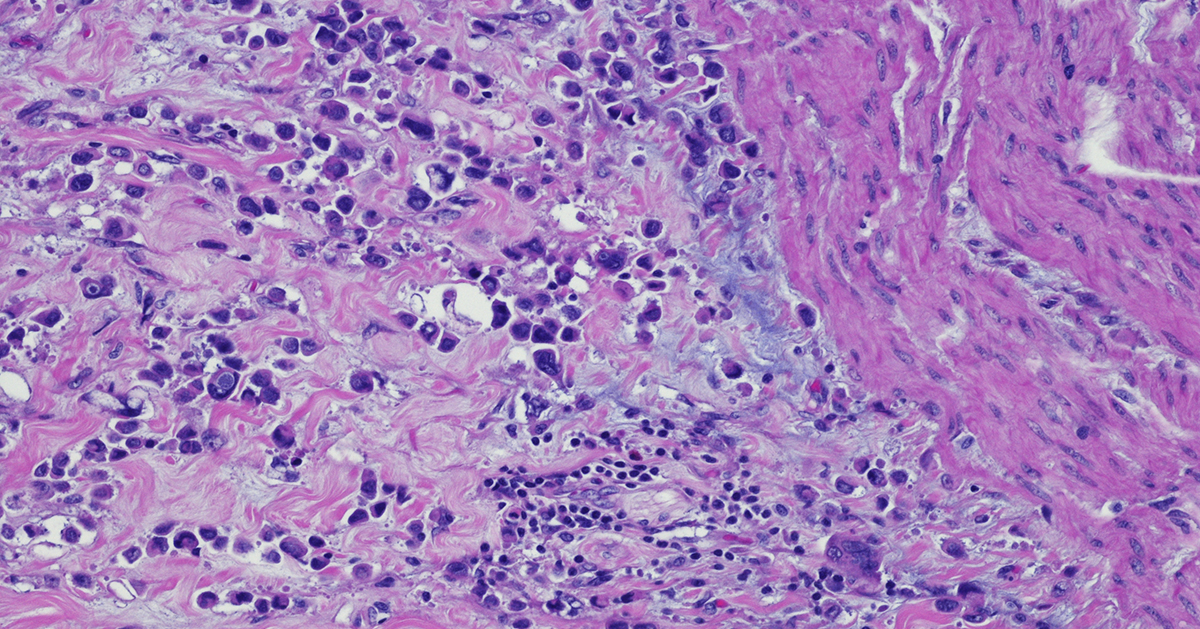
Immunohistochemistry: a Powerful Tool in Cancer Research
Immunohistochemistry (IHC) is a technique that originates in the early twentieth century but continues to be a valuable method that forms the backbone of molecular pathology. IHC is used for histological examination of tissues and specifically detects the presence of a molecule, such as a tumor antigen. IHC uses antibody-based labeling in which the primary antibody detects the target of interest and the secondary antibody detects the primary antibody which is linked to a molecule for microscopic visualization. Many different secondary antibody labeling modalities exist, including fluorescence, enzyme-mediated reactions and colloidal gold, and different labels are suited to specific microscopy platforms. Consider these five aspects of IHC as you implement this technique in preclinical cancer research: Quantitative measurements. IHC can be used as a qualitative measurement, but unlike many other visualization techniques, IHC can also be used as a quantitative measurement because antibodies that label specific parts of tissues or cells can be counted by a pathologist or with a computer-aided system. Developing robust validated quantitative IHC staining and visualization methods allows researchers to rely on the accuracy of this data. Customizable. IHC methods can be adapted to detect any cellular marker, given that a monoclonal antibody exists or can be made that specifically detects this marker. Validation of a new primary or secondary antibody also includes determining any off-target staining caused by these antibodies as this can be a critical determinant in the utility of an antibody. The ability to customize IHC in this manner is crucial to preclinical research that seeks to identify new biomarkers associated with tumor progression or immunotherapy efficacy. Flexibility. IHC can be used on almost any tissue type so long as it is processed correctly. Tissue samples from model animals as well as clinical biopsies can be fixed and sectioned in advance and stained at later times. Tumor microarrays (TMAs) can also be created and stained for evaluation of novel tumor markers or screening efficacy of drug candidates. This flexibility in sample type and handling highlights the overall utility of IHC. Automation. Currently, several different systems exist for automated IHC staining, and advances in digital analysis of IHC samples have allowed larger batches of clinical samples to be processed and evaluated. Comparison of automated IHC methods with manual methods has determined that these new approaches are accurate, sensitive, and reproducible. The big picture. Several methods exist for staining different targets in an IHC sample, and this allows scientists and clinicians to gain critical insights into which cells and molecules are present in the tumor microenvironment (TME), including levels of immune checkpoint molecules or infiltration of critical anti-tumor cell types. Consider how IHC can complement other techniques that look at the tumor microenvironment, including flow cytometry and RNAseq. IHC will continue to be a powerful tool in preclinical and clinical cancer research. Consider revisiting this classic technique as it has matured with the technological advances of the 21st century. Champions Oncology’s histology and immunohistochemistry services are custom developed and fully optimized to meet your needs in preclinical research. With industry leading pathology expertise and innovative automated technology, Champions provides you with the highest quality endpoints for your in vivo and ex vivo studies.

Uncovering Advantages and Disadvantages of Ex Vivo Culture in Preclinical Cancer Research
Ex vivo culture systems have been instrumental to preclinical oncology research because they enable researchers to study basic features of tumor cells and carry out large-scale screens of drugs or biologics. Ex vivo cultures do not fully recapitulate physiological conditions, although some advances have been made by developing three-dimensional (3D) culture methods. Here we highlight the advantages and disadvantages of using ex vivo cultures for preclinical oncology research. Cell culture: simple, inexpensive, but imperfect Ex vivo cell cultures of tumors were first developed as adherent two-dimensional (2D) monolayers that were grown in culture flasks or flat dishes. This approach is useful for studying tumors' basic cell biology and carrying out drug screens and preliminary toxicology and pharmacokinetic studies[1]. 2D cultures can be developed rapidly and cultured over longer periods of time using readily available tissue culture reagents. Unfortunately, 2D culture systems are limited in their applications because they do not accurately model the tumor microenvironment, and tumor cell morphology and function change under these culture conditions. Over the last several decades, 3D cultures have advanced significantly[2]. Several different systems now exist for 3D cultures, including liquid suspension cultures passaged on non-adherent plates, cultures grown in semi-solid substrates like soft agar or Matrigel, and cells grown on scaffolds like collagen. 3D culture systems better mimic characteristics of the tumor microenvironment, including cell-cell interactions, hypoxia, and reduced sensitivity to drug treatment[3]. (Visit our dedicated webpage to learn more about Champions' 3D tumor model offer) 3D cultures for drug discovery have become widely adopted over 2D models because they better predict in vivo efficacy and metabolic responses to drug treatments. A major advance in 3D tumor culture systems is the development of 3D spheroid and organoid cultures[4]. Spheroids are clusters of cells derived from tumors or other complex tissues and they cluster together through cell-cell adhesion. Organoids are more complex cell clusters derived from stem cells and can self-assemble and regenerate to form a smaller version of the original tumor or organ tissue. Tumor spheroids are widely used for drug screening and cytotoxicity assays and can be formed from dissociated tumor tissue or circulating tumor cells[5]. (Read our blog "Using 3D Ex Vivo Tumor Models for Oncology Research: An Expert Guide") Tumor organoids are even more accurate reflections of tumors growing under physiological conditions and are now widely used to compare the efficacy of standard-of-care treatments versus targeted therapies in addition to their use in drug screens[6]. Spheroids and organoids can undergo genomic editing as well, and this approach has been instrumental in identifying mutations that drive tumor growth or cause drug resistance[7-8]. (Read our blog "A Step-by-Step Guide to Design a 3D Tumor Model Co-Culture Study") Ex vivo + in vivo = the most effective strategy Animal models, especially patient-derived xenograft mouse models, are a critical bridge between preclinical studies and clinical trials. However, due to the lower costs and the compatibility with high throughput analysis, ex vivo tumor cultures are still the preferred method for ranking therapeutic agents, determining drug combination regimens, investigating mechanisms of action, and validating targets. Additionally, ex vivo tumor cultures are essential when studying certain hematological tumors, which are difficult to engraft and cannot be passaged in vivo because of their “liquid” nature. Both ex vivo and in vivo studies continue to work together to advance oncology research, but each year brings new advances in ex vivo culture systems that enhance their overall impact in predicting treatment responses.
Harnessing the Power of Oncolytic Viruses in the Fight Against Cancer
We typically consider viruses as infectious agents or vaccine vectors, which are non-replicating entities that express vaccine antigens. More recently, researchers have been working to develop oncolytic viruses, which are engineered to specifically infect and replicate in tumor cells. This targeted infection results in tumor cell lysis, activation of anti-tumor immune responses, and the release of new oncolytic virus particles that can infect other tumor cells. The concept of oncolytic viruses emerged in the early 20th century when scientists observed that a leukemia patient spontaneously went into remission for a brief time following an influenza virus infection. Early attempts to develop non-specific oncolytic viruses were plagued with complications related to poor efficacy and safety issues. As our understanding of targeted immunotherapy for the treatment of tumors has evolved, oncolytic viruses are gaining traction again and an oncolytic herpesvirus (talimogene laherparepvec or T-VEC) engineered to target metastatic melanoma was approved for clinical use by the FDA in 2015 [1]. Advantages of Oncolytic Viruses Oncolytic virus development has expanded significantly in the last two decades. Some of the most widely used viruses that are engineered to target tumors include adenoviruses, alphaviruses, herpes simplex viruses (HSV), rhabdoviruses, and vaccinia viruses. Viruses can be engineered to target tumor cells for lysis, such as replication-competent HSV-1 deletion mutants (e.g. thymidine kinase or ribonucleotide reductase mutants) that can only replicate in rapidly dividing tumor cells. Similarly, T-VEC is a modified HSV-1 virus with a gene deletion in ICP34.5, which allows for antiviral responses by normal cells but not tumor cells, thus allowing for tumor-specific virus-mediated destruction [2]. Oncolytic viruses can be modified to express immune molecules (e.g. TNF, IL-12, or chemokines) that promote proinflammatory responses and increase recruitment of macrophages and T cells for enhanced antitumor immunity in addition to oncolytic activity [3-4]. Oncolytic viruses can also induce apoptosis of tumor cells or enhance the uptake of chemotherapeutic agents [5]. Currently under pre-clinical investigation is a vaccinia virus carrying a TGFβRII inhibitor that has proved effective in causing tumor regression in mouse tumor models and shown an even greater effect when combined with checkpoint inhibitor therapy [6]. This approach overcomes the difficulties of targeting TGFβ, limiting side effects due to the targeting of non-tumor cells. Another promising approach currently being evaluated pre-clinically is a therapy using CAR-T and TCR-T cells infected with myxoma virus. This approach induces autosis and adaptive immunity in mouse models of tumors to restrain antigen escape [7]. Some oncolytic viruses can induce long-term immunity to tumors and prevent metastasis or the re-occurrence of these cancers, thus making them attractive therapeutic candidates. Clinical trials using oncolytic viruses are currently ongoing for numerous solid tumor types including glioblastoma, breast cancer, lung cancer, and bladder cancer [8]. Evaluating Oncolytic Viruses Oncolytic viruses are typically grown in tissue culture systems and are evaluated in vitro using a panel of tumor cell lines, which provides insight into tumor specificity and mode of action. These modified viruses can then be tested in a wide range of animal models, including immunocompetent mice, such as those used for syngeneic mouse tumor models, and immunocompromised mice, which include humanized mice that carry patient-derived tumor xenografts. Certain oncolytic viruses, such as vaccinia virus, require that researchers be vaccinated against this virus before laboratory handling, but in most cases, these viruses can be handled under BSL-2 conditions. Conclusions Despite great success in preclinical studies, translating oncolytic virus therapy to the clinic can be quite challenging. One major obstacle is the route of administration, with the oncolytic viruses needing to be injected directly into the tumor. While this can be simpler for superficial tumors like melanoma, it becomes much more complex to reach tumors that grow in deeper body organs. Many different varieties of oncolytic viruses are currently being evaluated preclinically or in clinical trials and the next decade promises further advances in this cutting-edge field.
Ultimate Guide: Designing a Mouse Clinical Trial & Data Analysis
Cancer is a complex disease, and developing novel, effective treatments requires testing in preclinical models. Mouse clinical trials represent a critical step in translating promising anti-cancer therapies from bench to bedside. When combined with advanced bioinformatics and analytics tools, mouse clinical trials are a powerful experimental approach to identifying target patient populations, planning cohort extensions, selecting the right combination drug, elucidating the mechanism of resistance, or identifying translational biomarkers [1]. However, conducting a successful mouse clinical trial requires expertise in designing the study, analyzing the data, and interpreting the results. In this blog post, we will guide you through the essential steps of designing a mouse clinical trial and analyzing the resulting data. This guide includes tips on powering the study, preparing the dataset, grouping responders vs non-responders, identifying biomarkers, and integrating proteomics in a multiomics model. Step 1: Powering the study In a mouse clinical trial, animals implanted with PDX models are surrogates of the patients whom PDX models were derived from. Before starting a mouse clinical trial, it is essential to determine the sample size required to achieve reliable and statistically significant results. It is important to enroll a large enough number of PDX models (which corresponds to the number of patients we would enroll) and enough animals per PDX model (so as not to lose a patient if we lose a mouse). Power analysis will estimate the minimum number of animals needed to detect a difference in tumor growth inhibition (TGI) between the treatment and control groups. Typically, researchers set the power at 80% and α at 0.05, which means that there is an 80% probability of detecting a significant difference between the two groups if one exists. It is also recommended to power mouse clinical trials according to the study goal and endpoint. For instance, a survival endpoint may require larger group sizes than a TGI endpoint. If the goal of the mouse clinical trial is to identify a novel biomarker of response, a larger number of models will need to be included in the design. Step 2: Select models Model selection is a crucial step when designing a mouse clinical trial and needs to be done by taking into account several parameters associated with the clinical history and molecular characteristics of the models so that the PDX model panel recapitulates the targeted patient population. Tumor indication and/or mechanism of action of the drug are usually the two key parameters that guide PDX model enrollment in a mouse clinical trial, and the deeper the information in terms of clinical classification and molecular profiling the more representative will be the PDX model panel [2]. (Read our blog: "How to Use Metadata as Your Model Selection GPS") Step 3: Preparing the dataset Once the study is completed, the next step is to prepare the dataset for analysis. This includes checking for outliers and removing/excluding missing data. One critical factor in analyzing mouse clinical trial data is the identification of PDX models with common response profiles. This is usually evaluated by a modified version of the clinical Response Evaluation Criteria in Solid Tumors (mRECIST). The response rate can be calculated by comparing the tumor volume at day 0 either with the volume corresponding to the best response measured on treatment or with the tumor volume measured on the final day of treatment. The threshold value can vary depending on the tumor type and drug mechanism of action. Therefore, it is crucial to validate the threshold on a panel of known drugs. Step 4: Grouping responders vs non-responders Once the threshold is established, the next step is grouping responders vs non-responders. Responders are mice that show a reduction in tumor volume or mass greater than or equal to the critical value, and non-responders are mice that show tumor reduction below the critical value. These groups are used to compare gene expression profiles and identify potential biomarkers of response [3]. Step 5: Identifying biomarkers Bioinformatics analysis can be used to identify biomarkers of response. One widely used approach is differential gene expression analysis (DGEA), which compares the gene expression profiles of responders and non-responders to identify genes that are differentially expressed. Another approach is differential gene set enrichment analysis (DGSE), which identifies functional gene sets that are enriched in the responders and non-responders. Partial least-squares (PLS) regression, a dimensional reduction method part of DIABLO multi-omics integration workflow [4], is commonly used to identify the most influential genes within a multi-omics gene network that can predict response. It is very important that mouse clinical trials are designed accordingly when performing multi-omics analysis as an endpoint. (Watch our webinar: "Identify MOA and Companion Biomarkers in Oncology using Multi-Omic Analyses") Step 6: Integrating proteomics for improved target validation and biomarker identification Proteomics approaches, such as mass spectrometry-based proteomics, can provide quantitative information on protein expression levels and post-translational modifications. Given the lack of correlation between protein abundance and RNA expression, integration of proteomics and phospho-proteomics data in a multi-omics model can greatly improve the accuracy of therapeutic target expression for model selection, provide a more comprehensive understanding of the molecular mechanisms underlying drug response, and provide an important additional molecular annotation for biomarker identification via single or multi-omics analysis [5]. (Read our blog: "4D Proteomics: Adding Dimension to Protein Detection") In this blog post, we have provided a step-by-step guide on how to design and analyze a mouse clinical trial, and shown how a successful mouse clinical trial requires careful planning and expertise in bioinformatics. Lumin Acuity offers tailored bioinformatics and computational biology solutions to support your mouse clinical trial planning and data analysis, accelerating decision-making and driving actionable results. Well-designed and executed mouse clinical trials can be instrumental in developing effective cancer therapies and improving patient outcomes.
Advancing Therapeutic Strategies for Myelodysplastic Syndrome
Myelodysplastic syndromes (MDS) are a group of hematological malignancies that manifest in hematopoietic stem cells and are caused by ineffective hematopoiesis.[1] Currently, MDS is defined as unexplained cytopenia combined with abnormalities in cell maturation leading to dysplastic features in >20% of myeloid cells.[2] MDS is one of the most frequently diagnosed malignancies in the United States and progresses to acute myeloid leukemia (AML) in 30% of patients.[2] While the initiation of primary MDS is not well understood, research suggests that somatic DNA injury, defective DNA repair, impaired immunological responses, and dysfunctional cell signaling play an important role in early stage MDS development.[3] Like other malignancies, MDS is positively selected for through gene mutations[4] with the average MDS patient carrying nine somatic mutations, such as TET2, TP53, and RUNX1.[4, 5] These mutations may not directly drive MDS, however, evidence indicates that mutations to genes involved with DNA methylation and RNA splicing greatly contribute to dysregulation of genes critical to hematopoiesis, such as GATA1, KLF1, and HOXA9.[6] MDS is classically characterized as hematopoietic stem cells (HSC) with clonal advantages due to somatic mutations or cellular dysfunction, discussed above. Studies have also identified the critical role of bone marrow microenvironments (BME) in MDS progression, particularly cytokine alterations and activities. Across several studies, high serum levels of TNF-α, TGF-β, IL-6, and IL-8[7] are present in MDS patient bone marrow (BM).[8] These proinflammatory molecules are typically associated with higher apoptotic rates, and in MDS patients, molecules such as TNF- α, correlate to poor MDS treatment performance.[7] Furthermore, studies have also identified the critical role of malignant clones in MDS pathogenesis. Mesenchymal stromal cells (MSCs) are a key component of the bone marrow that regulate hematopoiesis and have immunomodulatory properties. In contrast, dysplastic MSCs in the bone marrow can create a microenvironment that supports clonal expansion of malignant cells. Dysplastic MSCs from MDS or AML patients have phenotypic abnormalities, including aberration in secreted proteins and cell surface protein expression, as well as increased senescence and decreased survival.[9,10] Disrupted methylation profiles are observed in both MDS malignant clones and MDS-MSCs. These methylation changes impact multiple signaling pathways which create a chronically inflamed BME that is harmful to normal HSCs and supports the expansion of MDS clones.[11] While less understood, de novo MDS has been linked to chemotherapy, radiotherapy, and environmental carcinogens like benzene.[12] Therapy-related MDS (tMDS) is prevalent in 10-20% of patients 20 years after chemotherapy and/or radiation, and the World Health Organization (WHO) recognizes alkylating agents, like topoisomerase II inhibitors, as initiators of tMDS.[12] Due to the high incidence of MDS in elderly populations[13] with co-morbidities, treatment by bone marrow transplantation is often contraindicated and few therapeutic options exist. Patients with MDS are typically divided into two different categories: low-risk MDS and high-risk MDS. The division of MDS into these two groups is useful for researchers and clinicians developing therapies for this highly heterogeneic disease.[14] Low-risk MDS therapies target symptoms of cytopenia through the use of erythropoiesis stimulating agents (ESA), such as epoetin alfa and darbepoetin alfa.[15] The Food and Drug Administration (FDA) has also approved two hypomethylating agents (HMAs), 5-azacitidine and decitabine, for use in patients with low-risk and high-risk MDS. While both standards of care noncompetitively inhibit DNA methyltransferase (DNMT1) and promote hypomethylation of DNA to block DNA synthesis,[16] 5-azacitidine integrates with RNA and interferes with ribosomal assembly to also limit tumor protein synthesis.[16, 17] In 2023, the FDA and European Medicines Agency (EMA) also approved luspatercept, a recombinant fusion protein that enhances late-stage erythroblast differentiation, for low-risk MDS. Luspatercept indirectly moderates ineffective erythropoiesis by interacting with broad spectrum inhibitory signals, namely transforming growth factor-β (TGF- β) superfamily signaling.[18, 19] Downstream, signaling molecules, SMAD2 and SMAD3, negatively regulate TGF- β. Luspatercept works by binding to activin receptor type IIB on TGF- β which disrupts SMAD2 and SMAD3 signaling therefore improving erythropoiesis and boosting RBC production in MDS patients.[20, 21] As our understanding of MDS pathophysiology improves, more effective treatment options will become available. Researchers are actively evaluating the toxicity of allogeneic hematopoietic stem cell transplantation, a therapy with curative potential.[19] Given the nature of such treatment which involves conditioning chemotherapy, the risk of aplasia and other graft-versus-host disease need to be adequately assessed. Additional studies, such as a newly announced Montefiore Einstein Comprehensive Cancer Center clinical trial,[22] are investigating new therapies to develop novel therapeutic strategies.
A Multi-Omics Driven Approach for Advancements in Pancreatic Cancer
Pancreatic ductal adenocarcinoma (PDAC) is one of the most common and aggressive forms of pancreatic cancer that has remained difficult to diagnose early and treat successfully. PDAC has a five-year survival rate below 10% and is one of the leading causes of cancer death. Complete surgical resection is one of the few curative treatment modalities, and chemotherapy protocols have limited efficacy[1]. Unfortunately, most existing immunotherapy-based treatments are also associated with poor response rates[2]. Recent analyses of PDAC samples have provided critical insights into genetic alterations that drive tumorigenesis and have identified potential therapeutic targets. Here we highlight several of these key findings and how they may lead to advances in PDAC treatment. PDAC arises in the epithelial cells of pancreatic duct, or ductules, and is thought to progress in a manner like other carcinomas, in which the normal epithelial cells transition into pre-invasive pancreatic intraepithelial neoplasia lesions that eventually form invasive PDAC[3]. Most PDAC tumors carry somatic mutations in oncogenes, particularly KRAS, TP53, CDKN2A, and SMAD4[4]. KRAS mutation is the most frequent event in PDAC. The assumption that KRAS is an undruggable target based on extensive drug screens that showed in vitro inhibition but limited or no efficacy in animal models[5] is finally being challenged by the very promising results obtained by administration of Sotoresib (KRAS p.G12C inhibitor) in patients with advanced pancreatic cancer (NCT03600883), and by the identification of small-molecules that inhibit KRAS p.G12D in preclinical studies[6,7]. The need for druggable targets or biomarkers for PDAC has led to more innovative approaches to identify unique molecule attributes. A recent comprehensive proteogenomic characterization of PDAC pancreatic ductal tissues compared against paired normal adjacent tissue, and the findings validated known mutations in oncogenes[8]. This study also defined previously unknown genomic alterations and analyzed differences in protein expression and protein phosphorylation status between tissues. This comprehensive analysis identified a panel of proteins linked to early stage PDAC, and phosphoproteomic analysis identified several signaling pathways downstream of KRAS, including PI3K/AKT/mTOR and MAPK/ERK, that may be targeted by existing kinase inhibitors. The PAK1/PAK2 kinases were also identified as dysregulated in PDAC tissues and have the potential to be targeted therapeutically. Recent studies have also better characterized the tumor microenvironment (TME) of PDAC and how this may limit treatment efficacy[9]. PDAC tumors tend to be highly heterogenous with a dense stroma and disorganized blood vessels, which impair drug penetration. The TME is enriched for myeloid derived suppressor cells and regulatory T cells and displays a low mutational burden, thus making this a “cold” tumor immunophenotype that is resistant to existing immunotherapies. Current studies are examining immunotherapy or drug-based strategies to turn “cold” PDAC tumors into “hot” tumors that would be receptive to immune checkpoint blockade. Further advances in PDAC research will require similar extensive proteogenomic studies that better define dysregulated pathways that trigger early events in tumor formation and may function as early biomarkers for disease. The search for novel therapeutic targets or combinations of targets is also essential to improving the currently dismal array of treatment options for PDAC patients. Champions Oncology has 81 highly characterized Pancreatic Cancer Models available for preclinical studies, click below to learn more about how these models can propel your research.
4D Proteomics: Adding Dimension to Protein Detection
The field of oncology research has evolved significantly over the years, with new technologies constantly emerging to improve our understanding and treatment of cancer. One such technology is 4D proteomics, which utilizes four dimensions (retention time, mass, intensity, and ion mobility) to improve the detection of low-abundant peptides in complex biological samples. This powerful technique has proven to be particularly useful in personalized medicine and biomarker discovery for cancer research[1]. What is 4D Proteomics? Proteomics is the study of all proteins present in a given sample or organism. Traditional proteomic methods have typically focused on identifying and quantifying proteins in a sample based on their mass and abundance. However, this approach often falls short when it comes to detecting low-abundant peptides, limiting our ability to fully understand the complexity of biological systems. That's where 4D proteomics comes in. This technique adds an additional dimension (ion mobility) to the traditional three dimensions of mass, retention time, and intensity. Ion mobility measures the ability of a molecule to move through a buffer gas under an electric field, providing another valuable data point to improve peptide separation and identification[1]. The Benefits of 4D Proteomics in Oncology Research The use of 4D proteomics has greatly improved the sensitivity and specificity of peptide identification, making it particularly useful in oncology research where small changes in protein levels can have significant implications. The addition of ion mobility as a fourth dimension has also improved peptide separation and identification, making it easier to analyze complex samples such as blood or tissue from cancer patients. This is especially crucial in personalized medicine, where individualized treatments are tailored to a patient's specific cancer and its unique molecular characteristics[2]. Additionally, ion mobility helps differentiate each phosphopeptide isoform, accurately identifying the phosphorylation site on the peptide and quantitating each peptide form. This is critical for example in measuring certain kinases whose activity or substrate selectivity is controlled by phosphorylation at specific sites[3]. Conclusion In conclusion, the use of 4D proteomics has revolutionized the field of oncology research by improving our ability to detect low-abundant peptides and analyze complex samples. This technology has proven to be particularly valuable in personalized medicine and biomarker discovery for cancer research. As more studies continue to utilize 4D proteomics, we can expect further advancements in our fight against cancer.