

Gene copy number in a tumor cell is a significant indicator of the implication of a given gene in several oncogenic processes such as uncontrolled proliferation, elusion of programmed cell death, and resistance to treatments.
At Champions Oncology, gene copy number analysis is performed by using the EXCAVATOR2[1] tool on whole exome sequencing (WES) data generated to characterize our patient-derived xenograft (PDX) models. This tool allows for classifying each segmented region into five qualitative genomic states (two-copy deletion, one-copy deletion, normal, one-copy duplication, and multiple-copy amplification) and quantifying the number of chromosomal copies.
All our model characterization data can be explored in Lumin, a unique solution integrating Champions’ tumor model multi-omic data and public datasets in one accessible platform for model selection and data interpretation.
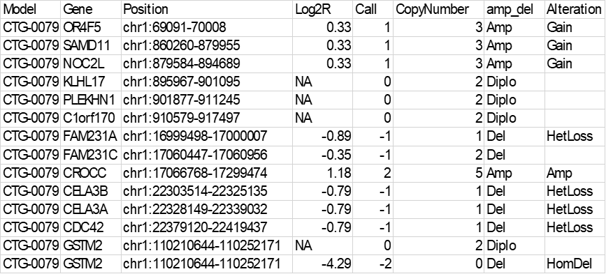
In Lumin, gene copy number analysis results are presented in the format shown in the example below:
Here we answer the most common questions about CNV data reporting to help you navigate WES data in our Lumin platform as well as interpret your own study data.
Q: What does Log2R mean and why sometimes is it marked as NA?
A: The Log2R (Log2 ratio) value represents the number of copies relative to the normal reference sample (NA12878). The EXCAVATOR2 algorithm used to calculate CNV uses a median normalization approach, with the log-transformed ratio (Log2R) being calculated from the window mean read count (WMRC) values of the test sample compared to the normal reference. When Log2R value is marked as NA, no significant copy number alteration was detected.
Q: What are the Call values and how are they defined?
A: The Call value is calculated using the FastCall algorithm[2] and classifies each segmented region as one of five possible states: 2 copy deletion= -2; one copy deletion= -1; normal= 0; one copy duplication= 1; and multiple copy amplification= 2.
Q: Are the copy number values the absolute or the relative copy numbers detected?
A: Copy number values represent the absolute copy number detected, which is derived from the Copy Number Fraction and is rounded to the nearest integer.
Q: Do “Amp” and "Del" always mean that there is a gain or loss in copy number? Or is this only the case if the “Alteration” column says “Gain” or “HomoDel”/ “Hetloss”?
A: The "amp_del" column definitions are derived from the Call values, whereas the "Alteration" column classifies the alteration based on the copy number detected. In some instances, there may be discordance between the copy number and Call (as shown for FAM231C gene in the example table above), as the two values are derived by approximation of continuous values. In this specific example, the conflict between the two annotations is indicative of the tumor gene copy number being between 1 and 2, suggesting the presence of both cells with 1 and cells with 2 copies of that genomic region. For additional investigation, we recommend looking at the continuous values in the raw data.
Q: What columns should I consider when I want to search for a model with an amplification/deletion for a certain gene?
A: We would first recommend using the "CopyNumber" column to identify models with an amplification or deletion of a specific gene. Once you have filtered the models based on this, you can then use the “Alteration” column to verify whether Call, copy number, and the “amp_del” column values are concordant.



