
Large-scale drug discovery increasingly depends on understanding mechanism, not just measuring viability. While high-throughput screening has traditionally relied on simple 2D systems, these models often fail to capture the biological complexity of human tumors or provide the mechanistic depth needed to make confident development decisions. At the same time, transcriptomic approaches have historically been too low-throughput to support large compound libraries.
In our AACR 2026 poster, we introduced scalable Chem-Seq and Functional-Seq platforms designed to close this gap. By combining high-throughput transcriptomic profiling with patient-derived organoid models, we can systematically map chemical and genetic perturbations at scale while preserving clinically relevant tumor biology.
Why transcriptomic profiling matters in drug discovery
Phenotypic screening alone rarely explains why a compound works or fails. Two agents may produce similar viability effects for completely different reasons, while others may appear inactive despite engaging their intended targets. Transcriptomic profiling addresses this challenge by capturing pathway-level responses and signatures of mechanism of action.
However, traditional RNA-seq workflows are difficult to scale. Sample preparation, library generation, and sequencing cost all become limiting factors. Our goal was to develop a workflow that preserves transcriptomic resolution while enabling tens of thousands of perturbations per week in relevant cancer models.
A scalable Chem-Seq workflow built for throughput
Our Chem-Seq platform is built around DRUG-seq™, a workflow optimized for bulk mRNA profiling directly from cancer cell lines or PDX-derived organoids in 384-well plates. Cells or organoids are plated, cultured, and treated with small-molecule compounds. Following treatment, a single-step lysis and barcoded oligo-dT capture allows mRNA from each well to be uniquely indexed in situ.
After reverse transcription, all wells are pooled, amplified, and processed through standard Illumina library preparation workflows. Libraries are sequenced to a depth sufficient to capture robust transcriptional signatures while maintaining scalability. This approach supports roughly 50,000 perturbations per week, spanning small molecules, and siRNAs.
The key advantage is efficiency. By eliminating many traditional bottlenecks, Chem-Seq enables rapid, reproducible transcriptomic profiling at a scale that aligns with modern screening demands.
From raw reads to mechanism-resolved insights
Chem-Seq data are processed to generate gene-by-barcode matrices, followed by normalization, batch correction, and dimensionality reduction. Principal component analysis and UMAP visualizations enable rapid triage of perturbation effects, while differential expression and pathway analyses reveal mechanism-associated signatures.
To further contextualize results, Chem-Seq signatures are compared against large public perturbation datasets using connectivity mapping approaches. This allows unknown compounds to be matched to known drugs, siRNA knockdowns, or CRISPR perturbations based on shared transcriptional responses, providing early insight into mechanism of action and potential off-target effects.
Anchoring chemistry with Functional-Seq
While chemical perturbations provide rich information, interpreting their biological meaning is often strengthened by genetic context. Functional-Seq integrates siRNA-mediated knockdown of drug targets directly into the same transcriptomic framework.
In this study, Functional-Seq was applied in non-small cell lung cancer PDX organoid models to generate loss-of-function signatures for selected targets. These genetic signatures serve as anchors for interpreting Chem-Seq profiles, enabling direct comparison between pharmacologic inhibition and genetic suppression of the same pathway.
Where chemical and genetic perturbations converge transcriptionally, confidence in on-target activity is strengthened. Where they diverge, additional biology, compensatory signaling, or off-target effects can be explored.
What the platform reveals in patient-derived organoids
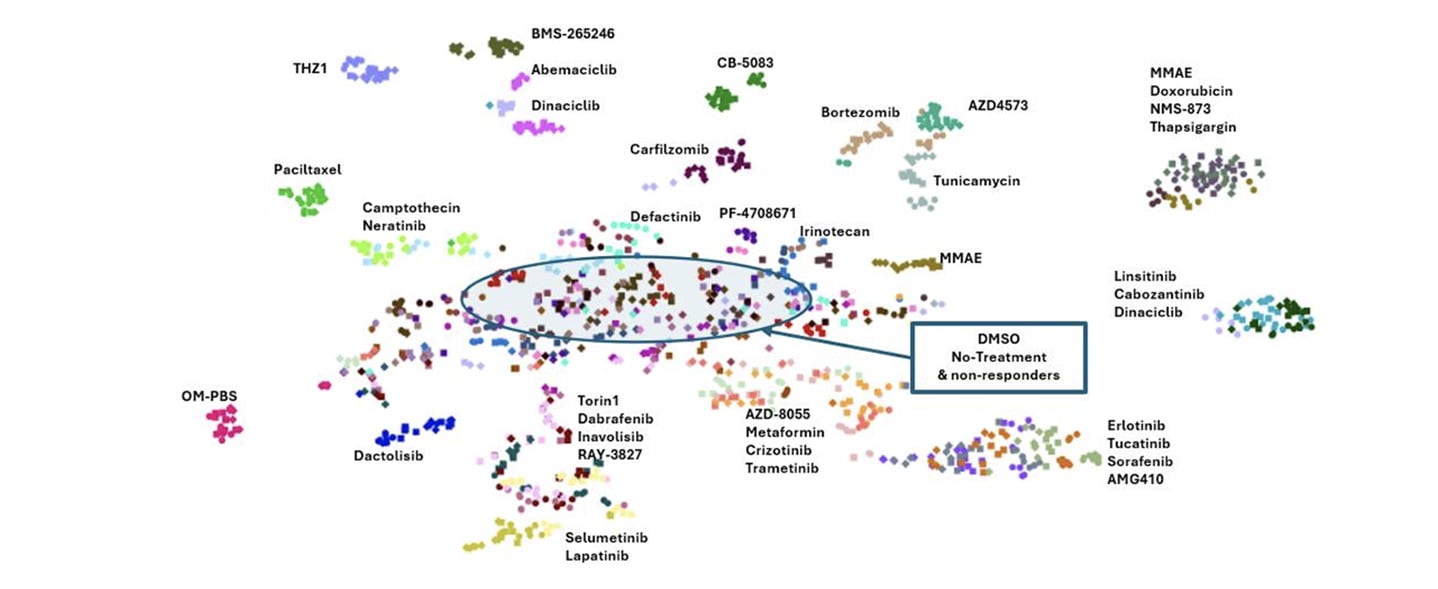
Using patient-derived organoids preserves key aspects of tumor heterogeneity and 3D architecture that are often lost in standard cell lines. Across the presented NSCLC PDX organoid models, Chem-Seq generated highly reproducible transcriptional signatures. Compounds targeting shared pathways clustered together, even when structurally distinct, while strongly cytotoxic agents formed separate transcriptional groups.
Dose-response profiling added an additional layer of insight. Increasing compound concentration revealed clear transitions between pathway-selective signatures and broader cytotoxic stress responses. This distinction is critical for prioritizing compounds that modulate their intended targets without inducing nonspecific toxicity.
Functional-Seq knockdowns recapitulated chemical inhibition for several pathways, demonstrating strong cross-method concordance at the gene and pathway levels. In contrast, targets that failed to produce robust genetic or pharmacologic signatures highlighted limitations related to knockdown efficiency or pathway redundancy.
Why integrated Chem-Seq and Functional-Seq matter
Together, Chem-Seq and Functional-Seq provide a scalable, mechanism-resolved framework for drug discovery in clinically relevant models. This integrated approach supports:
- Mechanism confirmation and target engagement assessment
- Rapid prioritization of compounds with clean, pathway-specific signatures
- Early identification of off-target or cytotoxic liabilities
- Improved interpretation of phenotypic screening data
Because the platform is compatible with large compound libraries and patient-derived organoids, it bridges the gap between discovery-scale screening and translational relevance.
Looking ahead
Following validation, this workflow is designed to scale further. Large-scale screening across diverse PDX organoid models and standard-of-care comparators will expand the reference space of transcriptomic signatures. Combined with machine-learning and generative AI approaches, these datasets can be used to predict compound response, guide structure-activity relationships, and even design novel compounds optimized for desired transcriptional outcomes.
By integrating high-throughput transcriptomics, genetic perturbation anchoring, and patient-derived 3D biology, Chem-Seq and Functional-Seq offer a powerful foundation for next-generation oncology drug discovery and precision medicine.
Want to explore the full data?
This blog highlights the overall strategy and key insights, but the complete poster includes detailed workflows, clustering analyses, concordance metrics, and pathway-level results.



